
Abstract
Accurate prediction of a particular cancer can be achieved by measuring multiplex biomarkers. Traditional methods for multi-biomarkers detection are either multi-spots assay with chip or multi-label assay with one detection spot. However, the detection throughput of these two approaches is limited by the substrate area and the numbers of available label respectively. To solve this problem, in the present study, an immunoassay was firstly prepared by combining multi-label strategy and multi-spot assay with a novel array electrode for simultaneous detection of six biomarkers for hepatocellular carcinoma (HCC). The detection throughput of the proposed method was doubled in comparison with traditional multi-spots assay (one target protein was detected on each analytic spot), which could greatly enhance the sensitivity and specificity of HCC diagnosis. This detection model may serve as the starting point for high throughput of multianalyte assay.
Similar content being viewed by others


Introduction
As cancer, in many cases, cannot be diagnosed and treated until cancer cells have already invaded surrounding tissues and metastasized throughout the body, which remains the leading cause of death in the population1,2. Therefore, early cancer detection is required for improving patient survival rate and, up to now, great efforts have focused on determination of biomarkers for early cancer detection3,4,5,6,7. Unfortunately, none of the currently available single biomarker could achieve 100% sensitivity and specificity8. For example, alpha fetoprotein (AFP) as a useful biomarker is often used to early diagnose hepatocellular carcinoma (HCC), but the false-negative rate with AFP level alone may be as high as 40% which resulted in miss diagnosis of HCC in early stage9,10. Another cancer biomarkers, such as lens culinaris agglutinin (LCA)-reactive fraction of AFP (AFP-L3), abnormal prothrombin (APT), α-L-fucosidase (AFU), des-γ-carboxy prothrombi (DCP) and γ-glutamyltranspeptidase (γ-GT), have been reported to be used in diagnosing HCC. However, the sensitivity and specificity of diagnosis was still unsatisfactory by only assessing single biomarker level11,12,13. Recently studies showed that accurate prediction can be achieved by measuring a panel of biomarkers of one cancer because the detection of multi-marker is complementary to each other for a particular cancer diagnosis14,15.Thus, it is necessary to pursue a simple method for diagnosis of HCC by combining with multi-markers in a clinical setting.
The dominant approach for multi-analyte detection is based on chip assay with the signal output according spatially distinct readouts of different bioactive species, which can also be referred to as multi-spot assay16,17,18,19,20. Although significant progress has been made in multianalyte assay using array chip, it is still a challenge to further raise throughput because of the number of detection spots is limited by the substrate area. More recently, researchers had developed the multiple-label strategy to realize the goal of one-spot multianalyte assay21,22,23. For example, Wang et al proposed a gensensor for simultaneous detection of multiple DNA targets based on different metal nanoparticles lables (ZnS, PbS, CdS) which could yield sensitive signals at different stripping voltammetric potentials with dissolving metal nanoparticles in nitric acid24. This method still encountered the predicament of high cost, long analysis time and complex detection process25. Therefore, in our pervious works, a novel sensitive approach has been proposed by labeling detection probes with various distinct redox probes (e.g thionine, ferrocene and anthraquinone-2-carboxylic acid) for simultaneously detection of multitargets on one-spot, which was much simpler, faster and more economical26,27,28,29. As our best knowledge, no attempt has hitherto been made to combine multi-label strategy and multi-spot assay to propose high throughput immunosersor for multiplex biomarker detection.
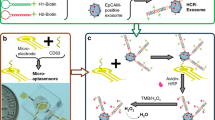
Herein, we developed a novel multianalyte immunoassay with high throughput for the simultaneous determination of six biomarkers combining multi-label strategy and multi-spot assay. The six biomarkers of AFP, AFP-L3, APT, AFU, DCP and γ-GT were simultaneously detected for the significant improvement in the sensitivity, specificity and detection efficiency of HCC early diagnosis. As show in Fig. 1, the newly designed array electrode was composed of three detection spots and a control one. Firstly, two different capture antibodies were immobilized on the each detection spot. Then the corresponding detection antibodies were labeled with the redox probes of thionine (TH) and anthraquinone-2-carboxylic acid (AQ), respectively. Based on the sandwiched immunoreactions, each individual protein could be detected with the distinct voltammetric peaks, whose position and size reflected the identity and level of the corresponding antigen (Fig. 1B). Thus, the six biomarkers could be detected simultaneously on one electrode with three analytical spots. Compared with traditional multi-spots assay (one target protein was detected on each detection spot), the detection throughput of the proposed method was doubled. Guided by this detection model, the detection throughput of the proposed immunosensor could increase three or four times more than the number of detection spots using the approach of simultaneous detection of three or four targets on one detection spot, which was reported in our previous work28,29. Thus, this detection model could serves as the starting point for high throughput of multianalyte assay. In addition, both of the graphene nanosheet/PtPd bimetallic nanocomposites (PtPd-GS NPs) and horseradish peroxidase (HRP) also labeled on the detection antibodies for a signal amplification to improve the sensitivity of the immunoassay.

Schematic diagrams of immunosensor array.
(A) preparation procedure of HRP and PtPd-GS labeled redox probe branched antibodies. (B) Schematic illustration of the stepwise immunosensor fabrication process and interaction of antibody-antigen.
Results
The structure of the novel array electrode
The novel array electrode for multi-spots assay was designed and the schematic diagram of its structure is shown in Fig. 1. One pair of the counter and reference electrodes (a platinum counter electrode, an Ag/AgCl reference electrode) are surrounded by four carbon working spots (2 mm in diameter. 1.5 mm edge-to-edge separation). Compared with the traditional chip electrode such as the disposable screen-printed electrode, this array electrode has the following three advantages: (1) Error of the detection results among different spots can be reduced because all the four working spots are exactly equal in space during the measurement process; (2) the cross-talk caused by diffusion of between adjacent spots can be effectively avoided due to the distance between spots is larger than the conventional array electrode; (3) the reproducibility of the novel electrode is improved compared with disposable screen-printed electrode because the novel electrode can be reused.
Characteristics of the different nanomaterials
Typical scanning electron microscopy (SEM) images are taken to observe the morphology of the as synthesized nanomaterials. Fig. 2A showed SEM image of the as-prepared pure graphene nanosheet (GS). Mono-layer sheets could be observed in most of regions, whereas restacked parts and wrinkles could also be seen, which could be attributed to electronic repulsion between the soft and flexible layers. After the generation of palladium nanoparticles (PdNPs) with the reducing of formic acid, rare highlights can be observed on the surface of GS shown in Fig. 2B. When several Platinum branches successfully grew from PdNP seed into bimetallic nanoparticles, very intensive highlights could be observed on the surface of GS (Fig. 2C). Alternatively, the morphologies of the Pd-GS and PtPd-GS also were evidenced by the transmission electron microscopy (TEM) image (Fig. 2D–F). It is observed that small Pd NPs supported on the surface of GS have a size of about 5–10 nm (Fig. 2D). The Pd NPs supported on GS were used as seeds to direct the dendritic growth of Pt upon the reduction of H2PtCl4 by ascorbic acid (AA) in an aqueous solution. Typical TEM images (Fig. 2E–F) revealed that Pt branches had grown from a Pd core into dendritic tendrils, indicating that PtPd-GS was successfully prepared. Furthermore, X-ray photoelectron spectroscopy (XPS) characterization was employed to further analyze the chemical composition of the PtPd-GS. XPS patterns of these hybrid nanomaterials show C1s, O1s and N1s signal characteristic of GS, Pt4f signal corresponding to the binding energy of metallic Pt (Fig. 2G) and Pd3d signal characteristic of metallic Pd (Fig. 2H), which further supported the conclusion that bimetallic PdPt NPs have been effectively assembled on the surface of GS.

SEM image of GS(A), GS-Pd NPs (B) and GS-PtPd NPs (C). TEM images of Pd-GN (D)and PtPd-GN at different magnifications (E and F). XPS of GS-PtPd NPs(G), Pd3d region (H).
Electrochemical characterization of the immunosensor
Electrochemical impedance spectroscopy (EIS) can provide detailed information about the preparation of the immunosensor and the immunobinding on the immunosensor. It is well known that the high frequency region of the impedance plot shows a semicircle related to the redox probe [Fe(CN)6]3−/4− and the semicircle diameter of EIS is equal to electron transfer resistance (Ret). The change of Ret value is associated with the blocking behavior of the modified layer on the electrode surface and reflected in the EIS as the change of the diameter of the semicircle at high frequencies. As seen in Fig. 3A, the EIS of bare electrode showed in curve a. After Au nanoparticles (AuNPs) were modified on the working electrode, a decrease in the Ret value was observed (curve b), since the excellent conductivity of AuNPs could promote electron transfer. Then the valve of EIS was increased when the mixture of capture antibodies was immobilized (curve c). Subsequently, BSA (0.25%) was immobilized to block the remaining active sites and a further of Ret value increased was obtained (curve d). The reason for this was antibody and BSA protein layers on the electrode would obstruct electron transfer. At last, the Ret value still increased after the prepared electrode was incubated with the mixture of antigens because the reaction of antigen-antibody occurred on the immunosensor surface.

(A) EIS obtained for different modified electrodes in 5.0 mM [Fe(CN)6]3−/4−: (a) bare electrode, (b) AuNPs/bare electrode, (c) Ab1/AuNPs/bare electrode, (d) BSA/Ab1/AuNPs/bare electrode, (e) Ag/BSA/Ab1/AuNPs/bare electrode. (B) Effect of the concentration of H2O2 on the response signals. The insert shows the DPV responds with different concentrations of H2O2. (C) Comparison of the electrocatalytic activities of different labeled probes by chronoamperometry: (a): HRP/Ab2-AQ/PtPd, (b): HRP/Ab2-AQ/PtPd-GS.
Optimization of the concentration of H2O2
The concentration of H2O2 played an important role in amplification signal. Therefore, it is necessary to investigate the effect of H2O2 concentration. After incubation of 2.1 AU L−1 DCP and 1.6 ng mL−1 AFP-L3, the immunosensor was investigated in 0.1 M HAc-NaAc (pH 5.5) with six concentrations of H2O2 (0.446, 0.609, 0.704, 1.15, 1.38, 1.59 mM) using differential pulse voltammetry (DPV) measurement. As seen from Fig. 3B, the reduction peak current increased rapidly with increasing the concentration of H2O2 and then tended to level off after more than 1.38 mM. Therefore, 1.38 mM H2O2 was adopted in the subsequent work.
Comparison of electrochemical behavior with different signal probes
To investigate the signal amplification of the proposed immunosensor, we chose γ-GT as an example and prepared two kinds of signal probes of HRP/Ab2-AQ/PtPd and HRP/Ab2-AQ/PtPd-GS. The same batch of immunosensors were incubated with 3.5 AU L−1 γ-GT and then incubated with different signal probes, respectively. These experimental procedures were performed with chronoamperometry measurement. As shown in Fig. 3C, the immunosensor incubated with HRP/Ab2-AQ/PtPd-GS probe (black line) was raised about 1.7 μA compared with the immunosensor incubated with HRP/Ab2-AQ/PtPd probe (red line). This may be attributed to the following reasons: (1) PtPd-GS as a carrier had more active sites and larger surface area than PtPdNPs, thus PtPd-GS could largely enhance the amount of immobilized the signal antibodies and effectively shuttle electrons between the electrode and redox probe; (2) In the presence of H2O2, a large amount of active PtPdNPs and HRP could greatly amplify the electrochemical response signal.
Cross-reactivity of immunoassay
It was necessary to investigate cross-talk and cross-reactivity in multiplexed immunoassay. Due to the distance between adjacent electrodes was larger than the diffusion distance of product, cross talk occurred among neighboring detection spot could been neglected. Therefore, we focused on investigating the cross-reactivity between different analytes. Firstly, the three detection spots modified with the capture antibodies of anti-AFP-L3 and anti-DCP were immunoreacted with the targets of (a) 2.4 ng mL−1 AFP-L3, (b) 3.2 AU L−1 DCP or (c) the mixture of 2.4 ng mL−1 AFP-L3 and 3.2 AU L−1 DCP, respectively. Then, the mixture of two signal probes (HRP/AFP-L3-Thi/PtPd-GS and HRP/DCP-AQ/PtPd-GS) was employed to perform the sandwich-type immunoreactions. Finally, the response signals of the three detection spots were measured by DPV (Fig. 4). The results suggested that the detection of multianalyte would not cause interference with each other and that cross-reactivity between the two analytes was negligible. These results also indicated that the proposed method with good specificity.

Typical DPV immunoassay signals for the investigation of cross-reactivity: (a) 2.4 ng mL−1 AFP-L3, (b) 3.2 AU L−1 DCP, (c) 2.4 ng mL−1 AFP-L3 and 3.2 AU L−1 DCP.
Multianalyte detection with the electrochemical immunosensor
The developed electrochemical immunosensor was evaluated by DPV in 2 mL HAc-NaAc with 1.38 mM H2O2. To assess the performance of the proposed immunoassay, we measured standard samples of antigen with different concentrations. It was seen that the amperometric responses increased with the increment of the corresponding antigens concentrations in 0.1 M HAc-NaAc solution (Fig. 5). The calibration plots showed good linear relationships in the ranges from 0.0320 to 3.20 AU L−1 for DCP, from 0.0240 to 2.40 ng mL−1 for AFP-L3, from 1.00 to 9.50 U L−1 for γ-GT, from 1.20 to 9.00 U L−1 for AFU, from 0.0250 to 5.00 ng mL−1 for AFP, from 0.0240 to 9.60 ng mL−1 for APT respectively. The detection limit was estimated 0.010 AU L−1 for DCP, 0.0080 ng mL−1 for AFP-L3, 0.33 for γ-GT U L−1, 0.40 U L−1 for AFU, 0.0080 ng mL−1 for AFP and 0.0082 ng mL−1 for APT respectively. The results identified that this method was efficient for sensitive electrochemical detection of six biomarkers of HCC.

DPV responses: The curves a, b, c, d, e in Fig. 5 (A) are the corresponding DPV after sandwich format immunoreaction with increasing DCP and AFP-L3 concentrations at 0.0320, 0.320, 1.60, 2.10, 3.20 AU L−1 and 0.0240, 0.240, 1.20, 1.60, 2.40 ng mL−1 respectively. The curves a′, b′, c′, d′, e′ in Fig. 5 (B) are the corresponding DPV after sandwich format immunoreactions with increasing γ-GT and AFU concentrations at 1.00, 2.00, 3.50, 6.00, 9.50 U L−1 and 1.20, 2.40, 4.00, 6.00, 9.00 U L−1 respectively. The curves a″, b″, c″, d″, e″ in Fig. 5 (C) are the corresponding DPV after sandwich format immunoreactions with increasing AFP and AFT concentration 0.0250, 0.250, 1.00, 2.50, 5.00 ng mL−1 and 0.0240, 0.240, 0.960, 4.80, 9.60 ng mL−1 respectively.
Clinical application
The proposed electrochemical immunosensor for clinical applications was investigated by standard addition methods in human serum. Different concentration of antigens were spiked into serum the samples. Experimental results were described in Table 1. The recoveries of them (95.0% to 105% for DCP, 95.0% to 103% for AFP-L3, 98.0% to 108% for γ-GT, 97.1% to 108% for AFU, 96.0% to 109% for AFP. 93.8% to 101% for APT) were acceptable, indicating that the proposed method provided a promising tool for determining biomarkers of HCC in real biological samples.
Discussion
A novel format of a high throughput immunosensor for simultaneous detection six biomarkers of HCC using a new array electrode was successfully demonstrated in this work. This design furnished the electrochemical immunosensor with three noticeable advantages: (1) A high throughput immunosensor was successfully proposed by combining multi-label strategy and multi-spot assay; (2) Electrochemical immunosensor array was an ideal alternative for low cost of the entire assay. Especially, in present work, the reuse of electrode can further reduce testing cost than a disposable electrode, which is also the best environmental option; (3) PtPd-GS NPs were used as bimolecular carriers in this system which could not only increase the antibody loading, but also amplify the signal for PtPd NPs catalytic activity. The method was used here to determinate six biomarkers, which had significant value in HCC diagnostics.
Methods
Chemicals and materials
Monoclonal primary antibody anti-AFP-L3, anti-APT anti-DCP, anti-AFU, anti-γ-GT (capture antibody), tracer secondary anti-AFP-L3, anti-APT anti-DCP, anti-AFU, anti-γ-GT (signal antibody), AFP-L3, APT, DCP, AFU and γ-GT standard solutions were purchased from Leibao Fu Co. (Shanghai, China). Monoclonal primary antibody AFP (capture anti-AFP) and tracer secondary anti-AFP (signal anti-AFP) and AFP standard solutions were also obtained from Biocell Co. (Zhengzhou, China). HRP, BSA, TH, AQ and chloroplatinic acid (H2PtCl6),gold chloride tetrahydrate (HAuCl4), potassium palladium (II) chloride (K2PdCl4) were obtained from Sigma Chemical Co. (St. Louis, MO, USA). Hydrazine, ammonia, formic acid, ascorbic acid (AA) and hydrogen peroxide (H2O2, 30%, w/v solution) were purchased from Chemical Reagent Co. (Chongqing, China), Graphite oxide (GO) was purchased from Nanjing Xianfeng nano Co. (Nanjing, China). N-hydroxyuccinimide (NHS) and N-(3-dimethylaminopropyl)-N-ethylcarbodiimeidehydride (EDC) were purchased from Shanghai Medpep Co.Ltd (Shanghai, China). Poly (vinylpyrrolidone) (PVP, MW = 40,000) was purchased from Beijing Chemical Reagent Co. (China). The acetate buffer solution (pH 5.5) was prepared with 0.1 M HAc-NaAc containing 0.1 M KCl. 0.1 M phosphate buffered solutions (PBS, pH 7.4) containing 10 mM Na2HPO4, 10 mM KH2PO4 and 2 mM MgCl2 was used throughout the experiment. All chemicals were analytical grade and used without further purification. All solutions were prepared with double-distilled water and stored in the refrigerator (4°C).
Apparatus
Cyclic voltammetric (CV), EIS and DPV measurements were carried out with a CHI 660C electrochemistry workstation (Shanghai CH Instruments, China). A conventional three-compartment electrochemical cell comprised a platinum wire auxiliary electrode, modified detection spot (Φ = 2 mm) and a saturated calomel reference electrode (SCE). The sizes and morphology of nanoparticles were estimated from scanning electron microscopy (SEM, S-4800, Hitachi, Japan). Transmission electron microscopy (TEM) images were obtained with a JEOL-JEM 2000CX transmission electron microscope (H600, Hitachi, Japan).The pH measurements were made with a pH meter (MP 230, Mettler-Toledo Switzerland) and a digital ion analyzer (Model PHS-3C, Da zhong Instruments, Shanghai, China).
Synthesis of PtPd-GS NPs
PtPd-GS NPs were obtained as following three steps30. Firstly, preparation of PVP-functionalized GS: 40 mg of PVP was added into 10 mL of homogeneous GO dispersion (0.25 mg/mL) with stirring for 12 h. Then, 3.5 μL of hydrazine solution (50% w/w) and 200 μL of ammonia solution (25% w/w) were added to the resulting dispersion. Following that the mixture was stirred for 1 h at 95°C. And then the stable black dispersion was centrifuged one time and dissolved in 1.25 mL of double-distilled water.
Secondly, preparation of Pd-GS NPs: Amounts of 500 μL PVP-functionalized GS aqueous solution were dissolved with double-distilled water, followed by the addition of 250 μL of K2PdCl4 (56.4 mM) and 0.35 mL of HCOOH. Then, the mixture was stored at room temperature until the Pd precursor was reduced completely. And the solution was centrifuged and dissolved in 3.8 mL of double-distilled water.
Thirdly, preparation of PtPd-GS NPs: the as-prepared suspension of Pd-GS NPs (3.8 mL), 250 μL of PVP aqueous solution (1 M) and 1.45 mL of AA aqueous solution (0.2 M) were added into a 10 mL flask. The mixture was heated to 90°C in air under magnetic stirring. Meanwhile, 0.5 mL of H2PtCl6 aqueous solution (0.1 M) was then rapidly injected into the flask by pipet. The reaction mixture was heated at 90°C in air for 3 h and then cooled to room temperature. The product was collected by centrifugation and washed several times with double-distilled water for further use.
Preparation of HRP and antibody multilabeled PtPd-GS NPs bioconjugates (Ab2 bioconjugates)
The antibodies and redox probes (antibody-redox probe) were conjugated by using EDC and NHS with the mole proportion of 4:1 as coupling agents. The antibody-redox probe conjugates were gently stirred for 12 h. Then, six kinds of antibody-redox probe conjugates were added into six beakers with 2 mL PtPd-GS NPs solutions respectively by incubation at 4°C for 8 h. The antibody-redox probe/PtPd-GS conjugates were collected by centrifugation and redispersed in PBS. Subsequently, HRP was dissolved in antibody-redox probe/PtPd-GS solution and incubated at 4°C for 4 h. Finally, after centrifugation, it was stored at 4°C for further use.
Fabrication of the immunosensor
The schematic illustration of the preparation procedure for HRP and antibody labeled the PtPd-GS NPs (AFP and APT as an example) as show in Fig. 1. Prior to detection, the electrode was immersed into HAuCl4 solution for electrochemical deposition under constant potential of −0.2 V for 30 s to obtain gold nanoparticles (AuNPs). Then three AuNPs modified spots were simultaneously incubated with different mixture solution (the mixture solution of anti-γ-GT and anti-AFU, the mixture solution of anti-DCP and anti-AFP-L3, the mixture solution of anti-AFP and anti-APT) respectively, which could be referred to as γ-GT and AFU detection spot, DCP and AFP-L3 detection spot and AFP and APT detection spot. The last one spot acted as blank control spot was incubated with mixture solution of anti-γ-GT and anti-AFU. Finally, the array electrode was incubated with BSA solution for 1 h at room temperature to eliminate nonspecific binding effects and block the remaining active groups.
The multianalyte detection was based on the typical procedure for sandwich-type immunoreactions. Firstly, the three analytic spots (γ-GT and AFU detection spot, DCP and AFP-L3 detection spot, AFP and APT detection spot) were incubated with the mixture solutions of corresponding antigens with different concentration at 37°C for 30 min respectively. Following that, three groups of the mixture solution of corresponding Ab2 bioconjugates were dropped on three analytic spots respectively and incubated for 30 min at 37°C. The blank control spot was incubated with BSA solution and then HRP/anti-γ-GT-AQ/PtPd-GS bioconjugates and HRP/anti-AFU-HT/PtPd-GS bioconjugates were dropped as blank control.
Experimental measurements
Electrochemical experiments were carried out in a conventional electrochemical cell, in which a modified working electrode, a Pt counter electrode and a SCE reference electrode were used. EIS of the electrode fabrication were performed in 5 mM [Fe(CN)6]3−/4− solution containing 0.1 M KCl. DPV was performed in 0.1 M HAc-NaAc (pH 5.5) containing 1.38 mM H2O2 to measure the amperometric responses of the immuosensor. The parameters applied were: 50 mV pulse amplitude, 50 ms pulse width, 0.2 s pulse period and voltage range from-0.6 to 0.1 V (vs. SCE).
References
Farazi, P. A. & DePinho, R. A. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat. Rew. Cancer 6, 674–687 (2006).
Wulfkuhle, J. D., Liotta, L. A. & Petricoin, E. F. Proteomic applications for the early detection of cancer. Nat. Rev. Cancer 3, 267–275 (2003).
Hazelton, W. D. & Luebeck, E. G. Biomarker-based early cancer detection: Is it achievable? Sci. Transl. Med. 3, 109fs9 (2011).
Vasilyeva, E. et al. Direct genetic analysis of ten cancer cells: tuning sensor structure and molecular probe design for efficient mRNA capture. Angew. Chem. Int. Ed. 50, 4137–4141 (2011).
Munge, B. S. et al. Nanostructured immunosensor for attomolar detection of cancer biomarker interleukin-8 using massively labeled superparamagnetic particles. Angew. Chem. Int. Ed. 50, 7915–7918 (2011).
Durner, J. Clinical chemistry: challenges for analytical chemistry and the nanosciences from medicine. Angew. Chem. Int. Ed. 49, 1026–1051 (2010).
Li, L. et al. Electrochemiluminescence energy transfer-promoted ultrasensitive immunoassay using near-infrared-emitting CdSeTe/CdS/ZnS quantum dots and gold nanorods. Sci. Rep. 3, 1529–1538 (2013).
Wagner, P. D., Verma, M. & Srivastava, S. Challenges for biomarkers in cancer detection. Ann. NY. Acad. Sci. 1022, 9–16 (2004).
Yao, D. et al. Abnormal expression of hepatoma specific g-glutamyl transferase and alteration ofg-glutamyl transferase gene methylation status in patients with hepatocellular carcinoma. Cancer 88, 761–9 (2000).
Cheng, H. T. et al. AFP-L3 in chronic liver diseases with persistent elevation of alpha-fetoprotein. J. Chin. Med. Assoc. 8, 310–317 (2007).
Ang, I. L. et al. Study of serum haptoglobin and its glycoforms in the diagnosis of hepatocellular carcinoma: a glycoproteomic approach. J. Proteome Res. 5, 2691–2700 (2006).
Shimada, M. et al. The role of des-γ-carboxy prothrombin levels in hepatocellular carcinoma and liver tissues. Cancer Lett 159, 87–94 (2000).
Kingsmore, S. F. Multiplexed protein measurement: technologies and applications of antibody arrays. Nat. Rew. Drug. Discov. 5, 310–321 (2006).
Hanash, S. M., Pitteri, S. J. & Faca, V. M. Mining the plasma proteome for cancer biomarkers. Nature 452, 571–579 (2008).
Stevens, E. V., Liotta, L. A. & Kohn, E. C. Proteomic analysis for early detection of ovarian cancer: A realistic approach? Int. J. Gynecol. Cancer 13, 133–139 (2003).
Lai, G., Yan, F., Wu, J., Leng, C. & Ju, H. Ultrasensitive multiplexed immunoassay with electrochemical stripping analysis of silver nanoparticles catalytically deposited by gold nanoparticles and enzymatic reaction. Anal. Chem. 83, 2726–2732 (2011).
Wu, Y., Xue, P., Kang, Y. & Hui, K. M. Paper-based microfluidic electrochemical immunodevice integrated with nanobioprobes onto graphene film for ultrasensitive multiplexed detection of cancer biomarker. Anal. Chem. 85, 8661–8668 (2013).
Wilson, M. S. & Nie, W. Multiplex measurement of seven tumor markers using an electrochemical protein chip. Anal. Chem. 78, 6476–6478 (2006).
Timm, C. & Niemeyer, C. M. On-chip protein biosynthesis. Angew. Chem. Int. Ed. 52, 2652–2654 (2013).
Deng, W. et al. Diagnosis of schistosomiasis japonica with interfacial co-assembly-based multi-channel electrochemical immunosensor arrays. Sci. Rep. 3, 1789–1794 (2013).
Qian, X. et al. Aptamer/Nanoparticle - based sensitive, multiplexed electronic coding of proteins and small biomolecules through a backfilling strategy. Chem. Eur. J. 16, 14261–14265 (2010).
Liu, G., Wang, J., Kim, J. & Jan, M. R. Electrochemical coding for multiplexed immunoassays of proteins. Anal. Chem. 76, 7126–7130 (2004).
He, S. et al. A graphene nanoprobe for rapid, sensitive and multicolor fluorescent DNA analysis. Adv. Funct. Mater. 20, 453–459 (2010).
Wang, J., Liu, G. & Merkoci, A. Electrochemical coding technology for simultaneous detection of multiple DNA targets. J. Am. Chem. Soc. 125, 3214–3215 (2003).
Osteryoung, J. G. & Osteryoung, R. A. Square wave voltammetry. Anal. Chem. 57, 101A–110A (1985).
Song, Z. et al. Horseradish peroxidase-functionalized Pt hollow nanospheres and multiple redox probes as trace labels for a sensitive simultaneous multianalyte electrochemical immunoassay. Chem. Commun. 46, 6750–6752 (2010).
Bai, L. et al. Simultaneous electrochemical detection of multiple analytes based on dual signal amplification of single-walled carbon nanotubes and multi-labeled graphene sheets. Biomaterials 33, 1090–1096 (2012).
Li, Y. et al. Simultaneous electrochemical immunoassay of three liver cancerbiomarkers using distinguishable redox probes as signal tags and gold nanoparticles coated carbon nanotubes as signal enhancers. Chem. Commun. 48, 537–539 (2012).
Zhu, Q., Chai, Y., Yuan, R. & Zhuo, Y. Simultaneous detection of four biomarkers with one sensing surface based on redox probe tagging strategy. Anal. Chim. Acta. 800, 22–28 (2013).
Guo, S., Dong, S. & Wang, E. Three-dimensional Pt-on-Pd bimetallic nanodendrites supported on graphene nanosheet: facile synthesis and used as an advanced nanoelectrocatalyst for methanol oxidation. ACS Nano 4, 547–555 (2010).
Acknowledgements
The authors are grateful for the National Natural Science Foundation of China (21275119, 21075100), the Ministry of Education of China (Project 708073), Specialized Research Fund for of the Doctoral Program of Higher Education (20100182110015) and the Natural Science Foundation Project of Chongqing (CSTC2009BA1003, CSTC2010BB4121, CSTC2011BA7003).
Author information
Authors and Affiliations
Contributions
Z.H.Y., Y.Q.C. and R.Y., designed the experiments manuscript, Z.H.Y. and Y.Z., performed experiments and wrote the main manuscript. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images in this article are included in the article's Creative Commons license, unless indicated otherwise in the image credit; if the image is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the image. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Yang, ZH., Zhuo, Y., Chai, YQ. et al. High throughput immunosenor based on multi-label strategy and a novel array electrode. Sci Rep 4, 4747 (2014). https://doi.org/10.1038/srep04747
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep04747
This article is cited by
-
High-Density Bulk Metallic Glasses and Their Composites for Kinetic Energy Penetrator Applications: Process, Structure and Properties
Transactions of the Indian Institute of Metals (2021)
-
Amperometric immunoassay for the tumor marker neuron-specific enolase using a glassy carbon electrode modified with a nanocomposite consisting of polyresorcinol and of gold and platinum nanoparticles
Microchimica Acta (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
