
Abstract
Surface exchange and oxygen vacancy diffusion dynamics were studied in double-perovskites LnBaCo2O5.5+δ (LnBCO) single-crystalline thin films (Ln = Er, Pr; −0.5 < δ < 0.5) by carefully monitoring the resistance changes under a switching flow of oxidizing gas (O2) and reducing gas (H2) in the temperature range of 250 ~ 800°C. A giant resistance change ΔR by three to four orders of magnitude in less than 0.1 s was found with a fast oscillation behavior in the resistance change rates in the ΔR vs. t plots, suggesting that the oxygen vacancy exchange diffusion with oxygen/hydrogen atoms in the LnBCO thin films is taking the layer by layer oxygen-vacancy-exchange mechanism. The first principles density functional theory calculations indicate that hydrogen atoms are present in LnBCO as bound to oxygen forming O-H bonds. This unprecedented oscillation phenomenon provides the first direct experimental evidence of the layer by layer oxygen vacancy exchange diffusion mechanism.
Similar content being viewed by others

Perovskite oxides exhibit a rich variety of interesting and important physical properties such as metal-insulator transition, giant magnetoresistance, spin blockade, etc. due to the complex and strongly correlated interactions among the charge, spin, orbital and lattice. Among them, the A-site ordered double perovskite cobaltates LnBaCo2O5.5+δ (LnBCO) (Ln = lanthanide, −0.5 < δ < 0.5) have recently attracted substantial attention not only due to their unusual electronic and magnetic properties as well as the metal-insulator transition but also their high electronic/ionic conductions for a variety of applications in solid oxide fuel cells, gas sensors, gas separation and permeation, electrochemical pumping systems, chemical energy storage systems and many others. The dynamics of oxygen surface exchange and the diffusion of oxygen vacancy1 become critical not only in governing the novel physical/chemical properties in the strong correlated interaction but also in determining the device performance such as energy efficiency, power densities of the batteries and fuel cells2 as well as the sensitivity and reliability of sensors. Therefore, it is a critical issue to understand the dynamics of their oxygen vacancy behavior both on the surface and in the bulk for multifunctional transition-metal oxides3,4. On the other hand, the recent researches on the highly epitaxial thin films of single-crystalline cobalt double-perovskites LnBaCo2O5.5+δ (LnBCO) (Ln = lanthanide, −0.5 < δ < 0.5) show various critical physical chemistry properties such as ultrafast oxygen diffusivity5 and high sensitivity to chemical environments6,7,8,9. Our recent research indicates that when the LaBCO films are exposed to a switching flow of oxidizing gas (O2) and reducing gas (the mixture of 4% H2 + 96% N2, which will be referred to as H2 for simplicity) in the temperature range of 250 ~ 800°C, their resistance R changes by three to four orders of magnitude in less than 0.1 s10. To understand the dynamic behavior of oxygen vacancy in the LnBCO systems, we carefully studied the resistance R and resistance change ΔR of thin epitaxial films of single-crystalline LnBCO (Ln = La, Er, Pr) as a function of the flow-time t of the oxidizing/reducing gases to find direct experimental evidence that oxygen/hydrogen atoms diffuse ultrafast through these films layer by layer via the oxygen-vacancy-exchange mechanism.
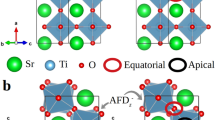
In general, a perovskite oxide ABO3 has a structure in which the BO2 layers alternate with the AO layers. In the A-site disordered perovskite LaSrCo2O6, there occurs only one kind of AO layers, i.e., the La0.5Sr0.5O layers in which the La3+ and Sr2+ ions are randomly distributed11. In the A-site ordered double-perovskite LaBaCo2O6, however, there occur two kinds of AO layers (i.e., LaO and BaO) leading to the repeat pattern of (CoO2)(LaO)(CoO2)(BaO) (Fig. 1a,d). The oxygen-deficient double-perovskites LaBaCo2O5.5 and LaBaCo2O5 have structures that have oxygen vacancies largely in the LaO layers (Fig. 1b,c). The oxygen vacancy in Fig. 1 (b) can be formed in the forms of order and disorder structures. Recently, highly epitaxial thin films of single-crystalline LnBCO (Ln = La, Er, Pr) were grown on (001) LaAlO3 by using pulsed laser deposition with a KrF excimer pulsed laser12,13,14,15,16. The oxidation and redox reactions of these films were monitored by measuring their resistance R under a switching flow of O2 and H2 as a function of the gas flow time t. The ac conductivity measurements indicate that both oxidation under O2 and reduction under H2 can occur at the temperature as low as ~200°C.

Schematic views of the A-site ordered cobalt double peroskite oxides.
(a) LaBaCo2O6. (b) LaBaCo2O5.5. (c) LaBaCo2O5. In (a)–(c) the atoms are distinguished by colored spheres: Co = cyan sphere, O = red sphere, Ba = blue sphere, La = orange sphere, the half-red/grey spheres in (b) indicate the oxygen site occupancy is half and oxygen vacancy = grey spheres in (c). (d) Sequence of the CoO2, BaO and LnO layers in LnBCO. The oxygen-vacancy-exchange diffusion between adjacent LnO and CoO2 layers is expected to be faster than that between adjacent CoO2 and BaO layers (indicated by large and small arrows, respectively) because more oxygen vacancies are present in the LnO layers.
Fig. 2 shows the R vs. t plots measured for single crystalline ErBCO thin films grown on (001) LaAlO3 at various temperatures under the switching flow of the reducing (H2) and oxidizing (O2) gases. The detailed R vs. t measurements for the reduction and oxidation cycles at various temperatures (Fig. 3a,b) show that at a given temperature the resistance change of the ErBCO films under oxidation is much faster than that under reduction. When the gas flow is switched from H2 to O2, the resistance drops down by a few orders of magnitude and this change depends sensitively on the temperature. The resistance under O2 or H2 is much higher at low temperatures than that at high temperatures. Below ~400°C the reduction under H2 occurs not in in one step, but in two steps (i.e., a very sharp increase followed by a gradual decrease). Under the repetitive switching of the redox environments, the reduction and oxidation processes of the ErBCO films are highly reversible as can be seen from Fig. 3c,d for the case of the redox reactions at 350°C.

Resistance vs. the gas flow time measured for single crystalline ErBCO thin films grown on (001) LaAlO3 at various temperatures under the switching flow of the reducing (H2) and oxidizing (O2) gases.

Resistance changes vs. the gas flow time, R vs. t, of an epitaxial ErBCO thin film.
(a) During the reduction (under H2) cycle at various temperatures. (b) During the oxidation (under O2) cycle at various temperatures. (c) At 350°C under the switching flow of the reducing (H2) and oxidizing (O2) gases. (d) Zoomed-in view of (c) for one set of reduction oxidation cycles.
The observed resistance of the LnBCO films under the flow of the redox gases can be understood by considering the average Co oxidation state. The resistance should be low when the average Co oxidation state is fractional (e.g., +3.5 and +2.5) because it signals the presence of mixed valence cobalt ions (e.g., Co3+/Co4+ and Co2+/Co3+, respectively) and hence the occurrence of either hopping with low activation energy or metallic conductivity. In contrast, the resistance of the LnBCO films should be high when the average Co oxidation state is an integer (e.g., +3 and +2) because electron-hopping is difficult in the case of single valence. The average Co oxidation state is +3.5 for LnBaCo2O6, +3 for LnBaCo2O5.5 and +2.5 for LnBaCo2O5. The Co3+/Co4+ and Co2+/Co3+ can be understood by doping levels of 0.5 holes and 0.5 electrons per Co3+ ion, respectively17. Our first principles density functional theory calculations for LaBaCo2O6 and various probable structures of LaBaCo2O5.5 and LaBaCo2O5(OH) show that hydrogen atoms are present in LnBCO as bound to oxygen forming O-H bonds. Then, the average Co oxidation state is +3 for LnBaCo2O5(OH) and +2.5 for LnBaCo2O4.5(OH) and +2 for LnBaCo2O3.5(OH)2. Fig. 3d shows that, as the gas flow is switched from O2 to H2, the resistance R of ErBCO increases to the maximum value and then decreases gradually to the equilibrium value. As the gas flow is switched from H2 back to O2, the resistance rapidly reaches its maximum value with the maximum rate of ~109 Ω/s and then quickly decreases to the stable value. These resistance changes are attributed to the average Co oxidation states on the surface of the films. In pure O2 environment, ErBCO can be fully oxidized to become ErBaCo2O6 with the Co average oxidation state of +3.5. As the gas flow changes from O2 to H2, ErBCO would be reduced to become ErBaCo2O5(OH) and then ErBaCo2O5.5 (by losing oxygen in terms of H2O) with average Co oxidation state of +3, which explains the sharp increase in the resistance. A further reduction under H2 would lead ErBCO to ErBaCo2O4.5(OH) and then ErBaCo2O5 with average Co oxidation state of +2.5, which explains the sharp decrease in the resistance.
At temperatures ranging from 230 to 400°C, the resistance change of the LnBCO films as a function of time in each oxidation cycle becomes oscillatory. As representative examples, the R vs. t and dR/dt vs. t plots obtained at 260°C for ErBCO are shown in Fig. 4a,b and those for PrBCO in Fig. 4c,d. The oscillations in the dR/dt curve occur in both the Co2.5+ → Co3+ and the Co3+ → Co3.5+ oxidation steps. Molecular dynamics studies on GdBCO18 indicate that the oxygen diffusion is much easier in the ab-plane than along the c axis. The oxygen vacancies in the LnO layers facilitate the oxygen ions to diffuse along the c-axis via the oxygen-vacancy-exchange diffusion mechanism between adjacent LnO and CoO2 layers and between adjacent BaO and CoO2 layers. In general, oxygen vacancies of LnBCO are present mainly in the LnO (Ln = La, Gd, Pr, Er, Nd) layers rather than in the BaO layers. Thus, in covering every c-axis length along the c-direction, the oxygen diffusion will experience two different rates as depicted in Fig. 1d. The hydrogen diffusion along the c-axis of LnBCO would experience a similar situation. Since the resistance of an LnBCO (Ln = Er, Pr) film is measured with the electrodes attached on the film surface, the measured resistance reflects the average Co oxidation state of the surface, which is affected by the hydrogen/oxygen diffusion from the surface to the inside of the film and by that in the opposite direction. The hydrogen/oxygen diffusion along the c-direction oscillates between a faster and a slower rate, so the average Co oxidation state on the film surface would oscillate hence leading to the oscillations of the dR/dt vs. t plot. The resistance oscillation in LnBCO requires the presence of the LnO and BaO layers with different extents of oxygen vacancies, so that LaSCO does not exhibit resistance oscillation (see Fig. S2 of SI). In the A-site ordered LnBCO, the LnO layer has more oxygen vacancies than does the BaO layer. Thus the change in the average Co oxidation state of a CoO2 layer would be greater when its vacancy-exchange involves the LnO layer than the BaO layer hence making the diffusion associated with the LnO layer more readily detectable than that with the BaO layer by resistance measurements. However, when the ionic radius difference between Ln3+ (e.g., Pr3+) and Ba2+ becomes smaller, more oxygen vacancies may exist in the BaO layer, so that the diffusion associated with the BaO layer can become visible. It is noted that the dR/dt oscillation of ErCBO is slightly different from that of PrBCO. Each dR/dt oscillation of PrBCO has two components (Fig. 4d), but that of ErBCO has mainly one (Fig. 4b). The larger oscillation peak of PrBCO is related to the diffusion through the PrO layer and the smaller one to that through the BaO layer. The oscillation peak of ErBCO is related to the diffusion through the ErO layer and the diffusion through its BaO layer is not observed most probably because the extent of its oxygen vacancy is very small. The atomic-exchange diffusion has been found for several pure metal surfaces such as Pt, Ir and Al19,20,21,22. However, there has been no direct experimental evidence for the vacancy-exchange diffusion taking place through the bulk of a crystalline material although this mechanism is often invoked to describe oxygen diffusion in various materials23. The resistance oscillation observed for LnBCO (Ln = Er, Pr) is the first experimental evidence for the occurrence of the oxygen-vacancy-exchange diffusion mechanism through the bulk of crystalline oxides.

(a, b) R vs. t and dR/dt vs. t plots for the epitaxial ErBCO films and (c, d) those for the epitaxial PrBCO thin films, taken for an oxidation cycle at 260°C. The insets in (a) and (c) show the zoomed-in view of the R vs. t plot in the regions of t = 11603 – 11608 and 7392 – 7412, respectively. The inset of (b) shows the Arrhenius plot, log D vs. 1000/T, based on the data of Table 1, with the negative and positive slopes indicated by solid and dashed lines, respectively.
The diffusion of hydrogen or oxygen along the c-direction through the lattice of ErBCO can be analyzed by Fick's second law24 for the one dimensional diffusion by considering a thin layer of thickness L equilibrated under a partial pressure P1 of redox gas. A sudden change of the partial pressure to another value P2 will cause the redox gas to diffuse eventually leading to the final state. This relaxation process can be monitored by resistance measurements. Given the carrier densities at the initial and final states as c1 and c2, respectively, the carrier density c(x, t) at the distance x from the center of the layer at the time t is related to the diffusion coefficient D of the redox gas (see SI). If the conductivity is dominated by one type of charge carrier, the mean conductance σm(x, t) is approximated as

where σ1 and σ2 are the conductance at the initial and final states, respectively and this expression is valid when t is small compared to the relaxation time  . Then, the conductance change Δσm during a short time interval Δt is given by
. Then, the conductance change Δσm during a short time interval Δt is given by

where  . We analyze the resistance oscillations observed during the oxidation cycle at various temperatures ranging from 240 to 400°C by employing Eq. (2) for each oscillation peak to deduce the associated relaxation times τ and subsequently the diffusion coefficients D by taking L = 2c where c is the c-axis lattice constant (see SI). Table 1 summarizes our results obtained at various temperatures ranging from 240 to 400°C.
. We analyze the resistance oscillations observed during the oxidation cycle at various temperatures ranging from 240 to 400°C by employing Eq. (2) for each oscillation peak to deduce the associated relaxation times τ and subsequently the diffusion coefficients D by taking L = 2c where c is the c-axis lattice constant (see SI). Table 1 summarizes our results obtained at various temperatures ranging from 240 to 400°C.
The dR/dt oscillations in ErBCO are absent above 320°C for the Co2.5+ → Co3+ oxidation process and also below 280°C for the Co3+ → Co3.5+ oxidation process (Table 1). This indicates that the Co2.5+ → Co3+ oxidation involves largely hydrogen diffusion and this diffusion becomes too fast above 320°C to cause dR/dt oscillations and that the Co3+ → Co3.5+ oxidation involves largely oxygen diffusion, which becomes too slow below 280°C to cause dR/dt oscillations. This interpretation is consistent with our observations that the diffusion coefficients D for the Co2.5+ → Co3+ oxidation (~10−2 cm2/s) are much greater than those for the Co3+ → Co3.5+ oxidation (~10−4–10−7 cm2/s) and the temperature-dependence of D is quite weak in the former oxidation process, but it is quite strong in the latter oxidation process. Thus our data show that the hydrogen diffusion is several orders of magnitude faster than the oxygen diffusion in ErBCO. The oxygen diffusion in ErBCO is faster than that found for other oxides by several orders of magnitude under similar temperatures (see SI). The oxygen diffusion in ErBCO is faster than that in YSZ, Gd:CeO2 and other similar perovskite oxides by several orders of magnitude25,26,27,28 while the hydrogen diffusion in ErBCO is comparable in rate to the silver diffusion in α-Ag2+δS and α-Ag2Te29.
In thermally activated electrical conduction, σ is related to the activation energy Ea as σ ∝ exp(-Ea/RT). Since σ is proportional to D, we plot log D vs. 1000/T using the data of Table 1 to determine Ea (see the inset of Fig. 4b). During the Co2.5+ → Co3+ oxidation process, the slope of the log D vs. 1000/T plot is slightly positive. This indicates that the ErBCO films behave as weakly metallic, which is consistent with our interpretation that the Co2.5+ → Co3+ oxidation largely involves hydrogen diffusion. Thus the activation energy for hydrogen hopping would be small (compared with that for oxygen hopping). During the Co3+ → Co3.5+ oxidation process, the log D vs. 1000/T plot changes from a negative slope above 280°C yielding Ea = 1.25 eV to a substantially positive slope below 280°C. This activation energy is much lower than that found for other oxide materials with disordered oxygen vacancies.
In summary, under the switching flow of O2 and H2 gases, the resistance change measured for epitaxial thin films of single-crystalline LnBCO (Ln = Er, Pr) as a function of the gas flow time t exhibit oscillations during the oxidation cycle under O2. This manifests that the ultrafast layer-by-layer exchange diffusion of O2 and H2 in LnBCO takes place by the oxygen-vacancy-exchange diffusion. Especially, the hydrogen ions can superfast diffuse in the ordered LnBCO systems may pave a new way for the studies of hydrogen in the ordered vacancy systems. These results suggest that the LnBCO systems are an excellent candidate for the development of low or intermediate temperature energy conversion devices.
References
Murch, G. R. & Nowick, A. S. Diffusion in Crystalline Solids [66–77] (Academic Press, Inc., 1984).
Wachsman, E. D. & Lee, K. T. Lowering the Temperature of Solid Oxide Fuel Cells. Science. 334, 935–939 (2011).
Burriel, M. et al. Anisotropic Oxygen Ion Diffusion in Layered PrBaCo2O5+δ . Chem. Mater. 24, 613–621 (2012).
Chen, Y. C., Yashima, M., Peña-Martínez, J. & Kilner, J. A. Experimental Visualization of the Diffusional Pathway of Oxide Ions in a Layered Perovskite-type Cobaltite PrBaCo2O5+δ . Chem. Mater. 25, 2638–2641 (2013).
Taskin, A. A., Lavrov, A. N. & Ando, Y. Fast Oxygen Diffusion in Perovskites by Cation Ordering. Appl. Phys. Lett. 86, 091910 (2005).
Kim, G. et al. Rapid Oxygen Ion Diffusion and Surface Exchange Kinetics in PrBaCo2O5+x with a Perovskite Related Structure and Ordered A Cations. J. Mater. Chem. 17, 2500–2505 (2007).
Kim, G. et al. Oxygen Exchange Kinetics of Epitaxial PrBaCo2O5+δ Thin Films. Appl. Phys. Lett. 88, 024103 (2006).
Liu, J. et al. Epitaxial Nature and Transport Properties in (LaBa)Co2O5+δ Thin Films. Chem. Mater. 22, 799–802 (2010).
Liu, J., Collins, G., Liu, M. & Chen, C. L. Superfast Oxygen Exchange Kinetics on Highly Epitaxial LaBaCo2O5+δ Thin Films for Intermediate Temperature Solid Oxide Fuel Cells. APL Mat 1, (2013) 031101.
Liu, J. et al. PO2 Dependant Resistance Switch Effect in Highly Epitaxial (LaBa)Co2O5+δ Thin Films. Appl. Phys. Lett. 97, 094101 (2010).
Senaries-Rodgiguez, M. A. & Goodenough, J. Magnetic and Transport Properties of the System La1-xSrxCoO3-δ (0 < x ≤ 0.50). J. Solid State Chem. 118, 323–336 (1995).
Liu, M. et al. Magnetic and Transport properties of Epitaxial (LaBa)Co2O5.5+δ Thin Films on (001) SrTiO3 . Appl. Phys. Lett. 96, 132106 (2010).
Ma, C. R. et al. Thickness Effects on the Magnetic and Electrical Transport Properties of Highly Epitaxial LaBaCo2O5.5+δ Thin Films on MgO substrates. Appl. Phys. Lett. 101, 021602 (2012).
Yuan, Z. et al. Epitaxial Behavior and Transport Properties of PrBaCo2O5 Thin Films on (001) SrTiO3 . Appl. Phys. Lett. 90, 212111 (2007).
Liu, M. et al. Giant Magnetoresistance and Anomalous Magnetic Properties of Highly Epitaxial Ferromagnetic LaBaCo2O5.5+δ Thin Films on (001) MgO. Appl. Mater. Interface. 4, 5524–5528 (2012).
Ma, C. R. et al. Magnetic and Electrical Transport Properties of LaBaCo2O5.5+δ Thin Films on Vicinal (001) SrTiO3 Surfaces. Appl. Mater. Interface. 5, 451–455 (2013).
Taskin, A. A., Lavrov, A. N. & Yoichi Transport and magnetic properties of GdBaCo2O5+x single crystals: A cobalt oxide with square-lattice CoO2 planes over a wide range of electron and hole doping. Phys. Rev. B 71, 134414 (2005).
Hermet, J., Geneste, G. & Dezanneau, G. Molecular Dynamics Simulations of Oxygen Diffusion in GdBaCo2O5.5 . Appl. Phys.Lett. 97, 174102 (2010).
Kellogg, G. L. &. Feibelman, P. J. Surface Self-Diffusion on Pt(001) by an Atomic Exchange Mechanism. Phys.Rev. Lett. 64, 3143–3146 (1990).
Chen, C. L. & Tsong, T. T. Displacement Distribution and Atomic Jump Direction in Surface Diffusion of Ir Atoms on the Ir(001) Surface. Phys. Rev. Lett. 64, 3147–3150 (1990).
Tsong, T. T. & Chen, C. L. Atomic Replacement and Vacancy Formation and Annihilation at Ir Surfaces. Nature. 355, 328–331(1992).
Feibelman, P. J. Diffusion path for an Al adatom on Al(001). Phys. Rev. Lett. 65, 729–732 (1990).
Ishigaki, T. et al. Diffusion of Oxide Ion Vacancies in Perovskite-Type Oxides. J. Solid State Chem. 73, 179–187 (1988).
Maier, J. Physical Chemistry of Ionic Materials. Maier, J. (ed.) 313 (Wiley, Chichester, 2004).
Minervini, L., Zacate, M. O. & Grimes, R. W. Defect Cluster Formation in M2O3-doped CeO2 . Solid State Ionics. 116, 339–349 (1999).
Bridges, C., Fernandez-Alonso, A. F., Goff, J. P. & Rosseinsky, M. J. Observation of Hydride Mobility in the Transition-Metal Oxide Hydride LaSrCoO3H0.7 . Adv. Mat. 18, 3304–3008 (2006).
Tarancón, A. et al. Layered Perovskites as Promising Cathodes for Intermediate Temperature Solid Oxide Fuel Cells. J. Mater. Chem. 17, 3175–3181(2007).
Maier, J. Mass Transport in the Presence of Internal Defect Reactions-Concept of Conservative Ensembles: I, Chemical Diffusion in Pure Compounds. J. Am. Ceram. Soc. 76, 1212–1217 (1993).
Maier, J. Physical Chemistry of Ionic Materials [318] (Wiley, Chichester, 2004).
Acknowledgements
This research was partially supported by the Department of Energy under DE-FE0003780, the Natural Science Foundation of China under 11329402 and the State of Texas through the Texas Center for Superconductivity at the University of Houston. Also, Mr. Shanyong Bao, Dr. Chunrui Ma and Mr. Xing Xu would like to acknowledge the support from the “China Scholarship Council” for the program of national study-abroad project for the postgraduates of high level universities at UTSA.
Author information
Authors and Affiliations
Contributions
S.B. had the sample preparation and conductivity measurements. C.M. had the microstructural characterizations, G.C., X.X. and E.E. assisted the sample preparation and conductivity measurements, C.C. designed, setup and supervised the research and discovered the exchange phenomena. Y.Z. made the targets for film fabrication. J.B. and M.W. conducted the modeling simulation, C.D. and Q.Z. assisted the data analysis. All authors discussed the results and commented on the manuscript written by S.B., C.C. and M.W.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images in this article are included in the article's Creative Commons license, unless indicated otherwise in the image credit; if the image is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the image. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Bao, S., Ma, C., Chen, G. et al. Ultrafast Atomic Layer-by-Layer Oxygen Vacancy-Exchange Diffusion in Double-Perovskite LnBaCo2O5.5+δ Thin Films. Sci Rep 4, 4726 (2014). https://doi.org/10.1038/srep04726
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep04726
This article is cited by
-
A-site double-lanthanide-doped La1−xPrxBaCo2O5+δ cathode materials for intermediate-temperature solid oxide fuel cells
Journal of Materials Science (2022)
-
Effect of Oxygen-deficiencies on Resistance Switching in Amorphous YFe0.5Cr0.5O3−d films
Scientific Reports (2016)
-
Manipulation of Optical Transmittance by Ordered-Oxygen-Vacancy in Epitaxial LaBaCo2O5.5+δ Thin Films
Scientific Reports (2016)
-
Anisotropic Strain Induced Directional Metallicity in Highly Epitaxial LaBaCo2O5.5+δ Thin Films on (110) NdGaO3
Scientific Reports (2016)
-
Gas Sensing Properties of Epitaxial LaBaCo2O5.5+δ Thin Films
Scientific Reports (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
