
Abstract
Iridates are of current great interest for their entangled spin-orbital state and possibly exotic properties. In this work, using density functional calculations, we have demonstrated that the hexagonal spin-chain materials Sr3MIrO6 (M = Ni, Co) are an iridate system in which the spin-orbit coupling (SOC) tunes the magnetic and electronic properties. The significant SOC alters the orbital state, the exchange pathway and thus the magnetic structure. This work clarifies the nature and the origin of the intra-chain antiferromagnetism of Sr3MIrO6 and well accounts for the most recent experiments.
Similar content being viewed by others


Introduction
Charge, spin and orbital states are often coupled in 3d transition-metal oxides due to their multiple degrees of freedom and electron correlation. These states are closely related to diverse material properties and functionalities, e.g., charge ordering, orbital ordering, spin-state and magnetic transition, metal-insulator transition, superconductivity, colossal magnetoresistance and multiferroicity. It is therefore very important to study those charge-spin-orbital states and their fascinating coupling for modeling and understanding of the abundant properties. This has formed a research stream in condensed matter physics over past decades, see e.g., a short review by Dagotto1. Very recently, research interest has been extended to 5d transition-metal oxides, which probably possess a significant spin-orbit coupling and provide an avenue to novel magnetic and electronic properties due to an entangled spin-orbital state.
In this respect, iridates are a representative example2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17. An octahedrally coordinated iridium ion normally has a large t2g-eg crystal-field splitting due to the delocalized character of its 5d electrons. The resultant low-spin state with only a t2g occupation makes an open shell Irn+ ion (e.g.,  for n = 4) behave effectively like p electrons (with an effective orbital momentum
for n = 4) behave effectively like p electrons (with an effective orbital momentum  ). As a result, an intrinsic strong spin-orbit coupling (SOC) splits the t2g levels into a lower
). As a result, an intrinsic strong spin-orbit coupling (SOC) splits the t2g levels into a lower  quartet and a higher
quartet and a higher  doublet. Then, for an Ir4+ constituent oxide, the half-filled
doublet. Then, for an Ir4+ constituent oxide, the half-filled  doublet may form, due to a moderate electron correlation, a novel
doublet may form, due to a moderate electron correlation, a novel  Mott insulating state2,3. It has been proposed that such a spin-orbital entangled state can bring about exotic properties, e.g., correlated topological insulator4,5, superconductivity6, Kitaev model7,8, Weyl semimetal9 and unusual magnetism10.
Mott insulating state2,3. It has been proposed that such a spin-orbital entangled state can bring about exotic properties, e.g., correlated topological insulator4,5, superconductivity6, Kitaev model7,8, Weyl semimetal9 and unusual magnetism10.
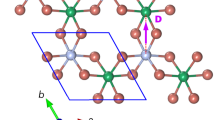
In this Report, we have studied the 3d–5d transition-metal hybrid material Sr3MIrO6 (M = Ni, Co) and find that the SOC has a significant impact on its magnetism by tuning its spin-orbital states and the Ir-M inter-orbital interactions. This system has a general chemical formula A3MM'O6 (A = Ca, Sr; M = 3d transition metal, M' = 3d, 4d, 5d transition metal) and displays an in-plane hexagonal structure and out-of-plane spin chains, see Fig. 1. Those quasi one-dimensional spin chains each consist of alternating face-sharing MO6 trigonal prisms and M'O6 octahedra. This system drew a lot of attention in the past decade18,19,20,21,22,23,24,25,26,27, because of its intriguing step-wise magnetization, significant Ising-like magnetism, thermoelectricity and multiferroicity. Sr3NiIrO6 and Sr3CoIrO6 also possess fascinating magnetism28,29,30,31. Owing to their complex temperature-dependent magnetic transitions, a standing issue is the nature of their dominant intrachain magnetism: either an intrachain ferromagnetic (FM) exchange28,29 or an antiferromagnetic (AF) coupling30,31 was proposed in previous studies. Moreover, the origin of the magnetism remains elusive. Therefore, Sr3NiIrO6 and Sr3CoIrO6 call for a prompt study to clarify the nature and origin of their intriguing intrachain magnetism. As seen below, we make a comparative study for Sr3NiIrO6 and Sr3CoIrO6, by carrying out a systematic set of electronic structure calculations. Our results consistently explain the experimental observations and settle the standing issue. In particular, we find that the SOC of the Ir4+ ion plays an essential role in determining the intrachain AF structure of Sr3MIrO6 (M = Ni, Co) by tuning the crystal-field level sequence and altering the Ir-M inter-orbital interactions. Therefore, Sr3MIrO6 is added to the iridate category which highlights the significance of the SOC.

Crystal structure plot of Sr3MIrO6 (a) projected onto the ab plane and (b) in a perspective view.
It has a hexagonal ab plane and quasi one-dimensional MIrO6 spin chains extending along the c-axis, in which the IrO6 octahedra and MO6 trigonal prisms are alternating.
Results
Crystal-field levels
We first carry out spin-restricted LDA calculations to estimate the crystal field splitting. The calculated DOS (density of states) results for Sr3NiIrO6 are shown in Fig. 2. The O 2p valence bands lie in between −7 and −2 eV. They have a significant covalency with the delocalized Ir 5d orbital and bring about the Ir 5d bonding state around −6 eV. A relatively weak Ni-O hybridization yields the Ni 3d bonding state in between −2 and −4 eV. Both the Ir 5d and Ni 3d antibonding states lie above −2 eV. For the Ir ion, its local octahedral coordination but a trigonal crystal field in the global coordinate system split the otherwise t2g triplet into the  doublet and the a1g singlet both of the concern. The eg doublet is far above them by 3 eV and is out of the concern. The a1g singlet can be written as 3z2 − r2, as the z-axis of the hexagonal lattice is along the [111] direction of the local IrO6 octahedra. Moreover, the
doublet and the a1g singlet both of the concern. The eg doublet is far above them by 3 eV and is out of the concern. The a1g singlet can be written as 3z2 − r2, as the z-axis of the hexagonal lattice is along the [111] direction of the local IrO6 octahedra. Moreover, the  doublet can be expressed as
doublet can be expressed as  and
and  , when the y-axis is set along the [
, when the y-axis is set along the [ ] direction of the local IrO6 octahedra (then the x-axis is uniquely defined and the xy is in the hexagonal ab plane). By integrating the DOS and determining the center of gravity for each eigen orbital within the antibonding energy range, we find that the
] direction of the local IrO6 octahedra (then the x-axis is uniquely defined and the xy is in the hexagonal ab plane). By integrating the DOS and determining the center of gravity for each eigen orbital within the antibonding energy range, we find that the  doublet is lower than the a1g singlet by 0.21 eV. Actually, the a1g-e'g level splitting is an important issue. The trigonal distortion of the IrO6 octahedron19, e'g-eg mixing32 and long-ranged crystal field due to the lattice anisotropy33 all contribute to the splitting. All these effects are properly included in the LDA calculation. Although the contribution of the lattice anisotropy is often quite complicate33, the a1g-e'g level ordering can be understood, as the elongation of the IrO6 octahedron along the [111] direction (the resultant O-Ir-O bond angle deviating from the ideal 90° by 5.4°28) raises a1g with respect to e'g and the e'g-eg mixing pushes e'g downwards32. For the Ni ion, its trigonal prismatic coordination produces the crystal-field level sequence of the Ni 3d electrons as 3z2 − r2/ xy, x2 − y2/xz, yz (0/0.57/0.76 eV). Furthermore, there are inter-site interactions between the Ir 5d and Ni 3d orbitals, see Fig. 2. The Ir a1g 3z2 − r2and Ni 3z2 − r2 electrons have a lobe pointing to each other and have a ddσ hybridization. The Ir
doublet is lower than the a1g singlet by 0.21 eV. Actually, the a1g-e'g level splitting is an important issue. The trigonal distortion of the IrO6 octahedron19, e'g-eg mixing32 and long-ranged crystal field due to the lattice anisotropy33 all contribute to the splitting. All these effects are properly included in the LDA calculation. Although the contribution of the lattice anisotropy is often quite complicate33, the a1g-e'g level ordering can be understood, as the elongation of the IrO6 octahedron along the [111] direction (the resultant O-Ir-O bond angle deviating from the ideal 90° by 5.4°28) raises a1g with respect to e'g and the e'g-eg mixing pushes e'g downwards32. For the Ni ion, its trigonal prismatic coordination produces the crystal-field level sequence of the Ni 3d electrons as 3z2 − r2/ xy, x2 − y2/xz, yz (0/0.57/0.76 eV). Furthermore, there are inter-site interactions between the Ir 5d and Ni 3d orbitals, see Fig. 2. The Ir a1g 3z2 − r2and Ni 3z2 − r2 electrons have a lobe pointing to each other and have a ddσ hybridization. The Ir  orbital has, via it's xz or yz component, a ddπ hybridization with the Ni xz/yz; and via it's xy or x2 − y2 component, a weak ddδ hybridization with the Ni xy/x2 − y2. Note that those crystal-field level sequences and the Ir-Ni inter-orbital hybridizations are crucial for understanding of the spin-orbital state and the intrachain magnetism in Sr3MIrO6, as seen below.
orbital has, via it's xz or yz component, a ddπ hybridization with the Ni xz/yz; and via it's xy or x2 − y2 component, a weak ddδ hybridization with the Ni xy/x2 − y2. Note that those crystal-field level sequences and the Ir-Ni inter-orbital hybridizations are crucial for understanding of the spin-orbital state and the intrachain magnetism in Sr3MIrO6, as seen below.

Partial density of states (DOS) of Sr3NiIrO6 in the nonmagnetic state calculated by LDA.
The octahedral Ir ion has a common large t2g − eg crystal field splitting of more than 3 eV; and in a trigonal crystal field (elongation of the IrO6 octahedron along the local [111] direction, i.e., the z-axis of the hexagonal lattice), the t2g splits further into a lower  doublet and a higher a1g singlet. The trigonal prismatic coordination produces the Ni 3d crystal-field level sequence (from low to high) as 3z2 − r2/xy, x2 − y2/xz, yz.
doublet and a higher a1g singlet. The trigonal prismatic coordination produces the Ni 3d crystal-field level sequence (from low to high) as 3z2 − r2/xy, x2 − y2/xz, yz.
Spin polarization and electron correlation
Then we perform spin-polarized LSDA calculations. We start with a FM or an AF state with the Ni2+ spin = 1 and Ir4+ spin = 1/2, but both calculations converge to a same FM metallic solution (not shown here). It has a total spin moment of 2.82 μB/fu, consisting of the local spin moments of 1.46 μB/Ni2+, 0.54 μB/Ir4+ and 0.11 μB/O. The Ni2+ ion has the electronic configuration  and the Ir4+ has a single t2g hole mostly on the a1g orbital [i.e.,
and the Ir4+ has a single t2g hole mostly on the a1g orbital [i.e.,  ]. The Ir-O and Ni-O covalencies bring about an appreciable spin moment of 0.11 μB on each oxygen. As the a1g orbital is a higher crystal-field level than the
]. The Ir-O and Ni-O covalencies bring about an appreciable spin moment of 0.11 μB on each oxygen. As the a1g orbital is a higher crystal-field level than the  , the a1g hole state allows a direct a1g electron hopping from Ni2+ to Ir4+ and this prompts the FM metallic solution.
, the a1g hole state allows a direct a1g electron hopping from Ni2+ to Ir4+ and this prompts the FM metallic solution.
The above LSDA metallic solution contradicts the experimental insulating behavior. In order to probe the electron correlation effect, we now carry out LSDA + U calculations. The electron correlation stabilizes the Ni2+ S = 1 state and the calculated spin moment of Ni2+ is enhanced to 1.68 μB. As seen in Fig. 3(a), the single Ir4+ t2g hole now fully occupies the down-spin a1g orbital. The Ir4+ spin moment is also increased to 0.64 μB. Owing to the correlation driven d-electron localization, the induced 2p spin moment on oxygen via the Ir-O and Ni-O hybridizations is reduced to 0.08 μB. Apparently, electron correlations make the Ir 5d and Ni 3d spin-orbital states fully polarized, thus giving an insulating solution. The tiny band gap is within the Ir t2g shell and it is due to a relatively weak electron correlation of delocalized 5d electrons. As Ir4+ has the single t2g hole on the down-spin a1g (3z2 − r2) orbital, the down-spin 3z2 − r2 electron of Ni2+ can hop forth and back. In order to maximize the local Hund exchange on the virtual Ni3+ ion in the excited intermediate state, the hopping 3z2 − r2 electron should be in the minority-spin channel, see Fig. 4 (b). Then this exchange mechanism gives the FM coupling between Ir4+ S = 1/2 and Ni2+ S = 1. Indeed, our LSDA + U calculation gives a total maximal spin moment of 3 μB/fu for this FM insulating state. Here we note that the Ir4+ a1g (3z2 − r2) orbital is orthogonal to the Ni2+ xz/yz and xy/x2 − y2 orbitals and hence there is no hopping between them to account for the magnetism. Moreover, the Ir4+eg empty bands are too high (see Fig. 2) to be relevant for the magnetic coupling. Therefore, with the crystal-field level diagrams depicted in Figs. 4(a) and 4(b), the electron correlation and inter-orbital hybridization give rise to the FM insulating solution.

The partial DOS of the Ir-5d a1g and  orbitals in Sr3NiIrO6 in (a) the FM state calculated by LSDA + U and in (b) the AF state by LSDA + U + SOC.
orbitals in Sr3NiIrO6 in (a) the FM state calculated by LSDA + U and in (b) the AF state by LSDA + U + SOC.
Taking the Ni2+ S = 1 as a reference, the Ir4+ ion has a down-spin a1g empty state in (a) but an up-spin  empty state (i.e., a complex orbital with lz = 1) in (b).
empty state (i.e., a complex orbital with lz = 1) in (b).

Schematic level diagrams of the Ir4+ 5d and Ni2+ 3d orbitals.
The down-spin 3z2 − r2electron mediates a FM coupling via a ddσ hybridization [(a) and (b)]. The up-spin xz/yz electrons mediate an AF coupling via a ddπ hybridization [(b) and (c)], in which the SOC splits the  doublet.
doublet.
Spin-orbit coupling
As 5d transition metals have an intrinsic strong SOC and particularly iridates are a representative example in this respect, now we are motivated to study the SOC effect by doing LSDA + U + SOC calculations. It is interesting to note that now we get an AF insulating solution with a small band gap of 0.15 eV, see Fig. 3(b). Particularly, this solution has the Ir4+ single t2g hole on the  orbital, in sharp contrast to the a1g hole state in the above LSDA + U FM insulating solution. The Ni2+ retains its configuration state and has a spin moment of 1.69 μB. Owing to the small crystal-field splitting of 0.19 eV between (xy, x2 − y2) and (xz,yz), a finite mixing between them due to the Ni SOC gives also a small orbital moment of 0.21 μB on Ni2+. The Ir4+ ion has now a spin moment of −0.44 μB. Moreover, the
orbital, in sharp contrast to the a1g hole state in the above LSDA + U FM insulating solution. The Ni2+ retains its configuration state and has a spin moment of 1.69 μB. Owing to the small crystal-field splitting of 0.19 eV between (xy, x2 − y2) and (xz,yz), a finite mixing between them due to the Ni SOC gives also a small orbital moment of 0.21 μB on Ni2+. The Ir4+ ion has now a spin moment of −0.44 μB. Moreover, the  doublet can form a complex orbital with lz = ±1. Then in the up-spin channel, the SOC lowers lz = −1 state and the lz = 1 state is pushed above the Fermi level by SOC and moderate electron correlation, determining the modest band gap and giving an orbital moment of −0.51 μB on Ir4+. Owing to the significant Ir-O covalency, both the spin and orbital moments are reduced from their respective ideal unit value. In the AF state of Ni2+ S = 1 and Ir4+ S = −1/2, the induced magnetic moment on oxygen gets tiny (only about 0.01 μB).
doublet can form a complex orbital with lz = ±1. Then in the up-spin channel, the SOC lowers lz = −1 state and the lz = 1 state is pushed above the Fermi level by SOC and moderate electron correlation, determining the modest band gap and giving an orbital moment of −0.51 μB on Ir4+. Owing to the significant Ir-O covalency, both the spin and orbital moments are reduced from their respective ideal unit value. In the AF state of Ni2+ S = 1 and Ir4+ S = −1/2, the induced magnetic moment on oxygen gets tiny (only about 0.01 μB).
Discussion
The above results indicate an interesting evolution of the intrachain magnetic structure, from the LSDA + U FM state to the LSDA + U + SOC AF state. It is ascribed to the SOC tuning orbital state of the Ir4+ ion. Although the a1g is a higher crystal-field level than the  by 0.21eV, the significant SOC of the Ir4+ ion (being about 0.5 eV) can well split the
by 0.21eV, the significant SOC of the Ir4+ ion (being about 0.5 eV) can well split the  doublet and eventually places the upper branch above the a1g (Fig. 4(c)). As a result, the single t2g hole of the Ir4+ ion lies in the
doublet and eventually places the upper branch above the a1g (Fig. 4(c)). As a result, the single t2g hole of the Ir4+ ion lies in the  state. Then, the up-spin xz/yz electrons of the Ni2+ ion can hop, forth and back, to the up-spin
state. Then, the up-spin xz/yz electrons of the Ni2+ ion can hop, forth and back, to the up-spin  empty state (i.e., the up-spin lz = 1 branch), giving rise to the AF coupling via the ddπ hybridization (Figs. 4(b) and 4(c)). Actually, using the orbital states depicted in Figs. 4(b) and 4(c), we also calculated the FM state. Our LSDA + U + SOC calculations find that the FM state is indeed less stable than the AF ground state by 67 meV/fu. Corresponding LSDA + U + SOC calculations, with Hund exchange parameter J = 0.9 eV for Ni 3d and J = 0.4 eV for Ir 5d, find that the AF ground state is more stable than the FM state by 63 meV/fu. In addition, the spin (orbital) magnetic moment changes within 0.1 (0.05) μB. Therefore, here the J parameter has an insignificant influence on the calculated results. As the SOC is intrinsic in iridates, the AF ground state is deemed reliable from the LSDA + U + SOC calculations, but the FM state seems fictitious from the LSDA + U calculations without inclusion of the SOC. Indeed, the AF ground state agrees with the most recent experiment30. Therefore, we can conclude that it is the significant SOC of the Ir4+ ion which tunes the spin-orbital states and Ir-Ni inter-orbital interactions and hence determines the AF structure of Sr3NiIrO6.
empty state (i.e., the up-spin lz = 1 branch), giving rise to the AF coupling via the ddπ hybridization (Figs. 4(b) and 4(c)). Actually, using the orbital states depicted in Figs. 4(b) and 4(c), we also calculated the FM state. Our LSDA + U + SOC calculations find that the FM state is indeed less stable than the AF ground state by 67 meV/fu. Corresponding LSDA + U + SOC calculations, with Hund exchange parameter J = 0.9 eV for Ni 3d and J = 0.4 eV for Ir 5d, find that the AF ground state is more stable than the FM state by 63 meV/fu. In addition, the spin (orbital) magnetic moment changes within 0.1 (0.05) μB. Therefore, here the J parameter has an insignificant influence on the calculated results. As the SOC is intrinsic in iridates, the AF ground state is deemed reliable from the LSDA + U + SOC calculations, but the FM state seems fictitious from the LSDA + U calculations without inclusion of the SOC. Indeed, the AF ground state agrees with the most recent experiment30. Therefore, we can conclude that it is the significant SOC of the Ir4+ ion which tunes the spin-orbital states and Ir-Ni inter-orbital interactions and hence determines the AF structure of Sr3NiIrO6.
Now we turn to Sr3CoIrO6. As this material has practically the same crystal structure as Sr3NiIrO628,30, both systems have many common features in the electronic and magnetic structures. The a1g singlet of the Ir4+ ion is higher than the  doublet in the crystal-field level diagram. Moreover, the high-spin Co2+ ion has the same crystal-field level sequence as Ni2+, but it now has one hole on the xy/x2 − y2 doublet (compared with Ni2+, see Fig. 4(b)). Our LSDA + U calculations give a FM metallic solution due to the 3z2 − r2 electron hopping and the 3/4 filled xy/x2 − y2 bands. Apparently, this solution contradicts the experimental AF insulating behavior30.
doublet in the crystal-field level diagram. Moreover, the high-spin Co2+ ion has the same crystal-field level sequence as Ni2+, but it now has one hole on the xy/x2 − y2 doublet (compared with Ni2+, see Fig. 4(b)). Our LSDA + U calculations give a FM metallic solution due to the 3z2 − r2 electron hopping and the 3/4 filled xy/x2 − y2 bands. Apparently, this solution contradicts the experimental AF insulating behavior30.
However, when we include SOC by doing LSDA + U + SOC calculations, we have obtained the correct AF insulating solution (see Fig. 5) in good agreement with the experiment30. The high-spin Co2+ ion (S = 3/2) has a spin moment of 2.66 μB. In its down-spin channel, the xy/x2 − y2 doublet form the complex orbitals (x2 − y2) ± ixy with lz = ±2 and the Co2+ SOC lowers the lz = 2 state but lifts the lz = −2 state. The electron correlation places the former at −1.5 eV and the latter at 2 eV. As a result, the Co2+ ion has also a huge orbital moment of 1.71 μB. In total, the Co2+ ion has the magnetic moment of 4.37 μB and it is firmly aligned, due to the SOC, along the hexagonal c-axis (i.e., a significant Ising-like spin system). Moreover, the Ir 5d states are almost the same as in Sr3NiIrO6: the Ir4+ SOC places the single t2g hole on the  doublet; the SOC and moderate electron correlation determine the small insulating gap within the Ir4+ t2g shell, see Fig. 5. The Ir4+ ion has the spin (orbital) moment of −0.39 (−0.47) μB and in total −0.86 μB. Using these spin-orbital states, our LSDA + U + SOC calculations find that the AF ground state is more stable than the FM state by 122 meV/fu, which changes little to 130 meV/fu when including Hund exchange J = 0.9 eV for Co 3d and J = 0.4 eV for Ir 5d.
doublet; the SOC and moderate electron correlation determine the small insulating gap within the Ir4+ t2g shell, see Fig. 5. The Ir4+ ion has the spin (orbital) moment of −0.39 (−0.47) μB and in total −0.86 μB. Using these spin-orbital states, our LSDA + U + SOC calculations find that the AF ground state is more stable than the FM state by 122 meV/fu, which changes little to 130 meV/fu when including Hund exchange J = 0.9 eV for Co 3d and J = 0.4 eV for Ir 5d.

The partial DOS of the Ir 5d and Co 3d eigen orbitals in AF Sr3CoIrO6 calculated by LSDA + U + SOC.
The high-spin Co2+ ion (S = 3/2) has the orbitals 3z2 − r2 (red curve) and  occupied (green curve) in its down-spin channel. The Ir4+ (S = −1/2) has the
occupied (green curve) in its down-spin channel. The Ir4+ (S = −1/2) has the  empty state (blue curve) in its up-spin channel.
empty state (blue curve) in its up-spin channel.
By looking at Figs. 4(b) and 4(c) and now having also one hole on the down-spin xy/x2 − y2 doublet for Co2+, we find that the net spin = 1 from xz/yz and the net spin = 1/2 from xy/x2 − y2 both contribute to the AF exchange with the net Ir spin = −1/2 from the  . The former is via a ddπ hybridization as in Sr3NiIrO6 and the latter a weaker ddδ one (which is missing in Sr3NiIrO6). This, together with the spin values (Co2+ S = 3/2 vs Ni2+ S = 1), qualitatively accounts for a higher stability of the AF ground state over the FM state in Sr3CoIrO6 than in Sr3NiIrO6, i.e., 122 vs 67 meV/fu. In addition, the ddδ hybridization results in the smaller spin and orbital moments of the Ir4+ ion in Sr3CoIrO6 (0.39 + 0.47 μB) than in Sr3NiIrO6 (0.44 + 0.51 μB). Note that all these results qualitatively explain the (slightly) higher intrachain AF transition temperature of 90 K in Sr3CoIrO6 than 85 K in Sr3NiIrO630. Moreover, the calculated magnetic moments of the significant Ising type, 4.37 μB/Co2+ and −0.86 μB/Ir4+, also agree reasonably well with the experimental ones of 3.6 and −0.6 μB in Sr3CoIrO630.
. The former is via a ddπ hybridization as in Sr3NiIrO6 and the latter a weaker ddδ one (which is missing in Sr3NiIrO6). This, together with the spin values (Co2+ S = 3/2 vs Ni2+ S = 1), qualitatively accounts for a higher stability of the AF ground state over the FM state in Sr3CoIrO6 than in Sr3NiIrO6, i.e., 122 vs 67 meV/fu. In addition, the ddδ hybridization results in the smaller spin and orbital moments of the Ir4+ ion in Sr3CoIrO6 (0.39 + 0.47 μB) than in Sr3NiIrO6 (0.44 + 0.51 μB). Note that all these results qualitatively explain the (slightly) higher intrachain AF transition temperature of 90 K in Sr3CoIrO6 than 85 K in Sr3NiIrO630. Moreover, the calculated magnetic moments of the significant Ising type, 4.37 μB/Co2+ and −0.86 μB/Ir4+, also agree reasonably well with the experimental ones of 3.6 and −0.6 μB in Sr3CoIrO630.
In summary, using density functional calculations including spin-orbit coupling (SOC) and electron correlation, we have demonstrated that the SOC of the Ir4+ ion plays an essential role in determining the antiferromagnetism of the hexagonal spin-chain system Sr3MIrO6 (M = Ni, Co) by tuning the crystal-field level sequence and altering the Ir-M inter-orbital interactions. The SOC splits the  doublet of the octahedral Ir4+ ion (
doublet of the octahedral Ir4+ ion ( ) in a trigonal crystal field and the single t2g hole resides on the
) in a trigonal crystal field and the single t2g hole resides on the  upper branch and gives rise to the antiferromagnetic superexchange. In absence of the SOC, however, the single t2g hole would occupy the a1g singlet instead, which would mediate an unreal ferromagnetic exchange due to a direct a1g hopping along the Ir-M chain. We also find that the Ni2+ and Co2+ ions are both in a high-spin state and moreover the Co2+ ion carries a huge orbital moment. This work well accounts for the most recent experiments and magnifies again the significance of the SOC in iridates.
upper branch and gives rise to the antiferromagnetic superexchange. In absence of the SOC, however, the single t2g hole would occupy the a1g singlet instead, which would mediate an unreal ferromagnetic exchange due to a direct a1g hopping along the Ir-M chain. We also find that the Ni2+ and Co2+ ions are both in a high-spin state and moreover the Co2+ ion carries a huge orbital moment. This work well accounts for the most recent experiments and magnifies again the significance of the SOC in iridates.
Methods
We have carried out density functional calculations, using the full-potential augmented plane wave plus local orbital code (Wien2k)34. We use the structural data of Sr3NiIrO6 measured by neutron diffraction at 10 K28 and of Sr3CoIrO6 at 4 K30. They have practically the same crystal structure: the Ir-O bondlength of 2.01 Å, the M-O 2.18 Å and the M-Ir 2.78 Å for M = Ni and Co; the small deviation of the O-Ir-O bond angle (from the ideal 90°) due to a small elongated trigonal distortion of the IrO6 octahedron, being 5.4° for M = Ni and 5.2° for M = Co. The muffin-tin sphere radii are chosen to be 2.8, 2.2, 2.2 and 1.5 Bohr for Sr, Ni/Co and Ir and O atoms, respectively. The plane-wave cut-off energy of 16 Ry is set for the interstitial wave functions and 5 × 5 × 5 k mesh for integration over the rhombohedral Brillouin zone. Using 7 × 7 × 7 k mesh (more than a doubling) gives practically the same results, with the total energy converging within 2 meV/fu. We employ the local spin density approximation35 plus Hubbard U (LSDA + U) method36 to describe the electron correlation of the M 3d and Ir 5d electrons, with the LSDA + U double counting correction made in a fully atomic limit36. The typical values, effective U = 2, 4 and 5 eV (Ueff = U − J with Hund exchange parameter J being set at zero) are used for the Ir 5d, Co 3d and Ni 3d states, respectively. Note that our key results –the coupled spin-orbital state and the magnetic ground state –are independent of the tested U values (1–3 eV for Ir 5d and 3–7 eV for Co/Ni 3d). To account for (near) degeneracy of the Ir 5d orbitals (and of M 3d orbitals as well), the SOC is included by the second-variational method with scalar relativistic wave functions. In order to probe diverse possible spin-orbital states and magnetic structures, we excess them in our calculations by setting their respective occupation number matrix and thus orbitally dependent potentials and then do self-consistent calculations including a full electronic relaxation. (Otherwise, some states of the concern or even the ground state cannot be achieved.) An advantage of this procedure is such that we can reliably determine the magnetic ground state by a direct comparison of the different states19,21.
References
Dagotto, E. Complexity in strongly correlated electronic systems. Science 309, 257–262 (2005).
Kim, B. et al. Novel Jeff = 1/2 Mott State Induced by Relativistic Spin-Orbit Coupling in Sr2IrO4 . Phys. Rev. Lett. 101, 076402 (2008).
Kim, B. J. et al. Phase-sensitive observation of a spin-orbital Mott state in Sr2IrO4 . Science 323, 1329–1332 (2009).
Shitade, A. et al. Quantum Spin Hall Effect in a Transition Metal Oxide Na2IrO3 . Phys. Rev. Lett. 102, 256403 (2009).
Pesin, D. & Balents, L. Mott physics and band topology in materials with strong spin–orbit interaction. Nat. Phys. 6, 376–381 (2010).
Wang, F. & Senthil, T. Twisted Hubbard Model for Sr2IrO4: Magnetism and Possible High Temperature Superconductivity. Phys. Rev. Lett. 106, 136402 (2011).
Jackeli, G. & Khaliullin, G. Mott insulators in the strong spin-orbit coupling limit: from Heisenberg to a quantum compass and Kitaev models. Phys. Rev. Lett. 102, 017205 (2009).
Chaloupka, J., Jackeli, G. & Khaliullin, G. Kitaev-Heisenberg Model on a Honeycomb Lattice: Possible Exotic Phases in Iridium Oxides A2IrO3 . Phys. Rev. Lett. 105, 027204 (2010).
Wan, X., Turner, A. M., Vishwanath, A. & Savrasov, S. Y. Topological semimetal and Fermi-arc surface states in the electronic structure of pyrochlore iridates. Phys. Rev. B 83, 205101 (2011).
Yin, W.-G. et al. Ferromagnetic Exchange Anisotropy from Antiferromagnetic Superexchange in the Mixed 3d–5d Transition-Metal Compound Sr3CuIrO6 . Phys. Rev. Lett. 111, 057202 (2013).
Haskel, D. et al. Pressure Tuning of the Spin-Orbit Coupled Ground State in Sr2IrO4 . Phys. Rev. Lett. 109, 027204 (2012).
Mazin, I. I., Jeschke, H. O., Foyevtsova, K., Valentí, R. & Khomskii, D. I. Na2IrO3 as a Molecular Orbital Crystal. Phys. Rev. Lett. 109, 197201 (2012).
Katukuri, V. M., Stoll, H., van den Brink, J. & Hozoi, L. Ab initio determination of excitation energies and magnetic couplings in correlated quasi-two-dimensional iridates. Phys. Rev. B 85, 220402 (2012).
Gretarsson, H. et al. Crystal-Field Splitting and Correlation Effect on the Electronic Structure of A2IrO3 . Phys. Rev. Lett. 110, 076402 (2013).
Li, Q. et al. Atomically resolved spectroscopic study of Sr2IrO4: experiment and theory. Sci. Rep. 3, 3073 (2013).
Cao, G. et al. Magnetism and electronic structure of La2ZnIrO6 and La2MgIrO6: Candidate Jeff = 1/2 Mott insulators. Phys. Rev. B 87, 155136 (2013).
Ou, X. & Wu, H. Coupled charge-spin-orbital state in Fe-or Co-doped Sr2IrO4 . Phys. Rev. B 89, 035138 (2014).
Niitaka, S., Yoshimura, K., Kosuge, K., Nishi, M. & Kakurai, K. Partially Disordered Antiferromagnetic Phase in Ca3CoRhO6 . Phys. Rev. Lett. 87, 177202 (2001).
Wu, H., Haverkort, M. W., Hu, Z., Khomskii, D. I. & Tjeng, L. H. Nature of Magnetism in Ca3Co2O6 . Phys. Rev. Lett. 95, 186401 (2005).
Choi, Y. J. et al. Ferroelectricity in an Ising Chain Magnet. Phys. Rev. Lett. 100, 047601 (2008).
Wu, H. et al. Ising Magnetism and Ferroelectricity in Ca3CoMnO6 . Phys. Rev. Lett. 102, 026404 (2009).
Agrestini, S. et al. Slow Magnetic Order-Order Transition in the Spin Chain Antiferromagnet Ca3Co2O6 . Phys. Rev. Lett. 106, 197204 (2011).
Kamiya, Y. & Batista, C. D. Formation of Magnetic Microphases in Ca3Co2O6 . Phys. Rev. Lett. 109, 067204 (2012).
Whangbo, M.-H., Dai, D., Koo, H.-J. & Jobic, S. Investigations of the oxidation states and spin distributions in Ca3Co2O6 and Ca3CoRhO6 by spin-polarized electronic band structure calculations. Solid State Commun. 125, 413–417 (2003).
Eyert, V., Laschinger, C., Kopp, T. & Frésard, R. Extended moment formation and magnetic ordering in the trigonal chain compound Ca3Co2O6 . Chem. Phys. Lett. 385, 249–254 (2004).
Vidya, R., Ravindran, P., Fjellvag, H., Kjekshus, A. & Eriksson, O. Tailor-made electronic and magnetic properties in one-dimensional pure and Y-substituted Ca3Co2O6 . Phys. Rev. Lett. 91, 186404 (2003).
Frésard, R., Laschinger, C., Kopp, T. & Eyert, V. Origin of magnetic interactions in Ca3Co2O6 . Phys. Rev. B 69, 140405(R) (2004).
Nguyen, T. & Zur Loye, H.-C. A Family of One-Dimensional Oxides: Sr3MlrO6 (M = Ni, Cu, Zn): Structure and Magnetic Properties. J. Solid State Chem. 117, 300–308 (1995).
Sarkar, S., Kanungo, S. & Saha-Dasgupta, T. Ab initio study of low-dimensional quantum spin systems Sr3NiPtO6, Sr3CuPtO6 and Sr3NiIrO6 . Phys. Rev. B 82, 23122 (2010).
Mikhailova, D. et al. Magnetic properties and crystal structure of Sr3CoIrO6 and Sr3NiIrO6 . Phys. Rev. B 86, 13409 (2012).
Zhang, G. R., Zhang, X. L., Jia, T., Zeng, Z. & Lin, H. Q. Intrachain antiferromagnetic interaction and Mott state induced by spin-orbit coupling in Sr3NiIrO6 . J. Appl. Phys. 107, 09E120 (2010).
Landron, S. & Lepetit, M.-B. Importance of t2g-eg hybridization in transition metal oxides. Phys. Rev. B 77, 125106 (2008).
Wu, H. et al. Orbital order in La0.5Sr1.5MnO4: Beyond a common local Jahn-Teller picture. Phys. Rev. B 84, 155126 (2011).
Blaha, P., Schwarz, K., Madsen, G., Kvasnicka, D. & Luitz, J. WIEN2k: An augmented plane wave plus local orbitals program for calculating crystal properties, Vienna University of Technology, Austria (2001).
Perdew, J. P. & Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 45, 13244–13249 (1992).
Anisimov, V. I., Solovyev, I. V., Korotin, M. A., Czyżyk, M. T. & Sawatzky, G. A. Density-functional theory and NiO photoemission spectra. Phys. Rev. B 48, 16929–16934 (1993).
Acknowledgements
This work was supported by the NSF of China (Grant No. 11274070), PuJiang Program of Shanghai (Grant No. 12PJ1401000) and ShuGuang Program of Shanghai (Grant No. 12SG06). X.O. was also supported by the Outstanding Doctoral Student Project of Fudan University.
Author information
Authors and Affiliations
Contributions
H.W. conceived the idea and designed the research. X.O. and H.W. performed the calculations. H.W. and X.O. prepared the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported license. The images in this article are included in the article's Creative Commons license, unless indicated otherwise in the image credit; if the image is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the image. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Ou, X., Wu, H. Impact of spin-orbit coupling on the magnetism of Sr3MIrO6 (M = Ni, Co). Sci Rep 4, 4609 (2014). https://doi.org/10.1038/srep04609
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep04609
This article is cited by
-
Intramolecular crossover from unconventional diamagnetism to paramagnetism of palladium ions probed by soft X-ray magnetic circular dichroism
Communications Chemistry (2020)
-
Spin–lattice and electron–phonon coupling in 3d/5d hybrid Sr3NiIrO6
npj Quantum Materials (2019)
-
Long-range magnetic interaction and frustration in double perovskites Sr2NiIrO6 and Sr2ZnIrO6
Scientific Reports (2014)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
