
Abstract
Low durability is the major challenge hindering the large-scale implementation of proton exchange membrane fuel cell (PEMFC) technology and corrosion of carbon support materials of current catalysts is the main cause. Here, we describe the finding of remarkably high durability with the use of a novel support material. This material is based on hollow carbon nanocages developed with a high degree of graphitization and concurrent nitrogen doping for oxidation resistance enhancement, uniform deposition of fine Pt particles and strong Pt-support interaction. Accelerated degradation testing shows that such designed catalyst possesses a superior electrochemical activity and long-term stability for both hydrogen oxidation and oxygen reduction relative to industry benchmarks of current catalysts. Further testing under conditions of practical fuel cell operation reveals almost no degradation over long-term cycling. Such a catalyst of high activity, particularly, high durability, opens the door for the next-generation PEMFC for “real world” application.
Similar content being viewed by others
Introduction
Fuel cells, in particular proton-exchange membrane fuel cells (PEMFC) represent a new energy technology with potential applications in the power demanding areas such as automobiles, portable electronic devices and distributed stationary power sources. This is because they possess advantages such as high power density, high efficiency and no pollution and are thus competitive with conventional energy conversion devices such as internal combustion engines and batteries1,2,3,4. However, there are still several challenges that hinder fuel cell commercialization, including insufficient durability/reliability and high cost and catalysts have been identified to be the main cause of these challenges5,6,7.
Within the last decades, much progress has been made for different kinds of anode and cathode catalysts. Alloyed Pt or non-Pt catalysts have been developed with the particular purpose of lowering the usage of Pt and thus the cost. Unfortunately, it is still very challenging to maintain or improve catalyst activity and durability when the Pt loading is reduced or eliminated. With the current state of technology, the state-of-the-art and the most practical electrocatalysts for PEMFC are still Pt based in the form of nanoparticles dispersed on carbon black supports5,6,7. However, these catalysts suffer from performance degradation during practical operation due to the high voltage, acidic and oxidation environment in PEMFC8. The corrosion of carbon support materials has been identified to be the major reason of the catalyst failure7,9, although other failure modes have also some contribution such as coarsening, dissolution, as well as poisoning of Pt particles10. For the cathode catalyst, in the presence of oxygen, oxidation of the carbon support can occur and result in the detachment of Pt particles and thus degraded fuel cell performance11. For the anode catalyst, the carbon support can also be oxidized in the situation of fuel (hydrogen) starvation12,13,14. As a result of these degradation processes, the stability of the Pt catalyst has still been short of the lowest 5,000-hour durability target for automotive applications, based on the testing of PEMFC vehicles monitored by the United States Department of Energy15,16. Therefore, under the strong driving force for fuel cell commercialization, the demand to replace current carbon supports using other intrinsically stable materials becomes essential and urgent. This is of significance not only for lengthening the operation life, but also for enhancing the reliability and reducing the total lifetime cost of PEMFC.
Considerable efforts have been made to explore stable alternatives for replacing the carbon materials (Vulcan XC-72R and Ketjen) currently used with fuel cell catalysts. These are based on some basic requirements, including high surface area, favored dispersion of catalytic metals, high oxidation resistance, high electrochemical stability under fuel cell operating conditions, as well as high electrical conductivity17,18. Challenges are that support materials can hardly meet all these requirements at the same time.
While graphene was studied only recently19,20,21,22, one-dimensional nanostructured carbon materials (carbon nanotubes-CNTs and carbon nanofibers-CNFs) have been receiving attention for a long time as catalyst supports because of their unique structure and properties23,24,25,26. It has been concluded that the structure defects play an important role in improving catalytic activity. However, the corrosion of carbon materials always initiates at defect sites. The influence of structure defects on the durability of electrocatalysts supported by CNTs and CNFs is still an open question17. Enhanced durability of Pt/CNTs was observed for CNTs with a good graphitic structure, which is attributed to the strong resistance of CNTs to corrosion and the specific interaction between Pt nanoparticles and CNTs (the delocalized electrons of CNTs and Pt d-electrons)18,27,28. However, the dispersion of Pt on the support with a high degree of graphitization is poor and nonuniform, because of few defects available for the nucleation of Pt. Further shortcoming of CNTs is their inferior dispersion in the solutions for preparing both catalyst and catalyst ink for membrane electrode assembly (MEA), because of their long lengths of tens or even hundreds of micrometer. To overcome this shortcoming, short CNTs of a few hundred nanometers long have to be prepared by solid state cutting or direct synthesis29,30.
Conductive doped diamond is intrinsically attractive for application as a durable catalyst support because of its specific properties, such as an extremely wide potential window, a very low background current and in particular a high chemical and dimensional stability31,32,33. However, there are still some problems with doped diamonds as electrocatalyst supports: the low conductivity, the low surface area and the poor dispersion of the catalytic metals. In addition, it is still difficult to realize a homogeneous and controllable boron doping level in diamond powders34,35.
Mesoporous carbon materials have a large surface area with three-dimensionally interconnected mesopores and have been extensively studied as catalyst supports36,37,38. The supported Pt and Pt alloy catalysts have shown excellent performance in electrode reactions, mainly due to the high and uniform dispersion of catalytic metals and enhanced mass transfer39,40,41,42. In order to improve its electrical conductivity and stability, mesoporous carbon has to be treated by high-temperature annealing and catalytic graphitization43,44. However, such treatment strategies have to be implemented at the expense of the porosity and specific surface area, making it difficult to deposit metal nanoparticles.
To address the issue of carbon oxidation, great effort has been placed in recent years on developing noncarbon materials for catalyst supports, including nitrides, carbides, mesoporous silicas, conducting polymers and metal oxides (for references, see a review paper7). Although some progress has been made, significant drawbacks still exist for these supports when compared to carbon materials, mainly including lower electronic conductivity, higher solubility in acidic environment and lower specific surface area. Some noncarbon materials have intrinsic inertness to high temperatures and corrosive electrochemical environments, significantly improving the stability of the supports as well as their supported catalysts. But, others, such as WC, are easier to oxidize at oxygen electrode potentials.
In this study, we report a new support material: carbon nanocages (CNCs), a kind of spherical nanocarbon with a graphitic shell and a hollow interior. This material was prepared by spray pyrolysis of iron carbonyl and carbon precursor. Graphitization was varied by changing pyrolysis temperature and in-situ N doping was achieved by using a N-containing carbon precursor. Emphasis was placed on the effects of graphitization and N doping on deposition of Pt particles and the performance of the resultant catalyst for H2 oxidation reaction (HOR) and oxygen reduction reaction (ORR). As will be shown, the Pt catalyst supported on N-doped CNCs with good graphitic structure exhibits remarkable catalytic activity and stability. Under the electrochemical conditions similar to practical fuel cell operation, the catalyst demonstrates a constant activity for both HOR and ORR during extended periods of potential cycling.
Results
Physical characteristics of hollow CNCs
The as-prepared samples consisted of nanoparticles with an Fe-phase core and a graphitic shell. The overall size of the particles and the thickness of the shell appeared to increase with increasing pyrolysis temperature. In order to obtain hollow CNCs, the solid core had to be removed. Conventional method was boiling in concentrated nitric acid, but removal efficiency was low. We propose a heat treatment method with the inclusion of sulfur powder. Almost all particles were emptied with a minimal loss of carbon material and a minimal destruction of the graphitic shell (Supplementary Figures S1–S3 in Supplementary Information).
The TEM images of hollow CNCs with graphitic shells after solid core removal are shown in Figure 1. As can be seen, with increasing preparation temperature, the particle size and the shell thickness increased obviously. The size of the hollow cages prepared at 700°C was 10–50 nm in outer diameter with the shell thickness being 2–5 nm and that for CNCs prepared at 1,000°C was 20–50 nm with the shell thickness of 3–10 nm. But, for the 1,500°C sample, the particle size was 50–100 nm and the thickness of the graphitic shell was 5–15 nm. Moreover, HRTEM images showed that the shell was composed of well-developed graphitic layers with a spacing of 0.34 nm (Figure 1b, d, f).

TEM and HRTEM images of CNCs prepared at different temperatures.
(a, b) 700°C, (c, d) 1,000°C, (e, f) 1,500°C.
Figure 2a shows the XRD patterns of different hollow CNC samples and only diffraction peaks for graphite can be observed. The diffraction peaks at 2θ = 26.1° are attributed to the diffraction from the (002) planes of graphite. The peak intensity became obviously stronger with increasing temperature. For CNCs prepared at 1,500°C, the peaks at 2θ = 42.6° and 51° correspond to the (100) and (004) planes of graphite. The graphitization of the hollow CNCs can be further characterized by Raman spectroscopy and TGA.

Physical characteristics of hollow CNCs prepared at different temperatures.
(a) XRD patterns, (b) Raman spectra, (c) TGA curves and (d) XPS spectra (compositions are in at.%).
Raman spectroscopic spectra (Figure 2b) exhibit two peaks at approximately 1,350 and 1,581 cm−1. The peak at 1,581 cm−1 can be identified as the G peak of perfect crystalline graphite arising from the zone-center E2g mode and the peak at 1,350 cm−1 as the D peak assigned to the A1g zone-edge phonon induced by the disorder in the graphite lattice45,46. These results demonstrate that the degree of graphitization improved with increasing temperature.
TGA results (Figure 2c) show that from ambient temperature to about 400°C, CNCs prepared at 700°C underwent a little oxidation with weight loss of about 10%. Significant oxidation reaction took place at 400–550°C. However, the CNCs prepared at 1,000 and 1,500°C exhibited no weight loss up to about 500°C and strong weight loss occurred at about 600°C. These results confirm again that CNCs prepared at higher temperatures had a higher degree of graphitization.
After complete oxidation, the residual weight was 3.97%, which could be attributed to the formation of iron oxide. After the process of Fe removal, there was still a little amount of iron remaining in the sample. Such iron was usually tightly entrapped in thick graphitic shells and difficult to be removed. During the heating at low temperatures up to 400°C in TGA experiments, such iron underwent little oxidation due to the protection of graphitic shells. As a result, there was no weight gain in this temperature range.
X-ray photoelectron spectroscopy (XPS) was conducted to further study the surface composition of the hollow CNCs. Typical XPS survey scan and the surface composition of the samples are shown in Figure 2d. The peak at about 284.8 eV was attributed to C1s and the O1s peak existed at about 533 eV, indicating that the present CNCs consisted of mainly carbon and oxygen. The observed asymmetric tailing for C1s was a result of the presence of some functionalization of the surfaces of the CNCs. It should be noted that the CNCs synthesized using pyridine as carbon precursor contained a little amount of nitrogen. The atomic content of N in CNCs prepared at 700°C and 1,000°C was 2.2 and 0.96%, respectively. The main reason for the low contents of N might be the high temperatures used for pyridine pyrolysis and the subsequent heat treatment, perhaps leading to C-N bond breaking and elimination of some doped nitrogen, respectively. The hollow CNCs prepared at high temperature also contained a trace amount of sulfur, which might come from the S used in the process of Fe core removal.
Pt/CNC catalysts and their electroactivities
Figure 3 shows typical TEM, HRTEM micrographs and XRD patterns of different Pt/CNC catalysts. For Pt/CNC(700) and Pt/CNC(1000), Pt nanoparticles were fine and uniformly dispersed on the outer surfaces of CNCs. There was no evidence for the deposition of Pt particles inside the CNCs, probably because the CNCs were closed with a multi-layered graphitic structure as shown in Figure 1. Statistic estimation of more than two hundreds of Pt particles revealed that the average size of the Pt particles was about 2.25 and 3.25 nm for Pt/CNC(700) and Pt/CNC(1000), respectively (Supplementary Figure S4a, b). However, the Pt nanoparticles supported on CNCs prepared at 1,500°C dispersed non-uniformly and aggregated forming bigger particles. The mean particle size of Pt was about 7.25 nm. The XRD patterns (Figure 3f) illustrated the peak for graphite at about 2θ = 26° and the peaks for Pt(111), Pt(200), Pt(220) at 2θ = 39.8°, 46.2° and 67.5°, respectively. The peak intensity became stronger with increasing CNC preparation temperature, indicating an increase of Pt particle size. The weight percentages of Pt in Pt/CNC(700) and Pt/CNC(1000) were estimated from TGA experiments to be 43.22 and 44.37%, respectively (Supplementary Figure S5a, b), which are close to the nominal Pt loading of 45 wt.%.

TEM images and XRD patterns of different Pt/CNC catalysts.
(a,b) Pt/CNC(700), (c,d) Pt/CNC(1000), (e) Pt/CNC(1500), (f) XRD patterns of Pt/CNC.
The initial cyclic voltamograms of different catalysts are shown in Supplementary Figure S6. As can be seen, Pt/CNC(700) demonstrated stronger hydrogen desorption and adsorption peaks and thus a higher activity than Pt/CNC(1000) and Pt/CNC(1500). When compared with the commercial catalyst, the present catalyst using CNCs as supports exhibited an outstanding electrochemical activity.
The electrochemical surface area (ECSA) for a catalyst can be estimated from the following equation47,48:

where Q is the charge of hydrogen desorption, m the quantity of Pt on electrode and β the charge required to oxidize a monolayer of H2 on bright Pt (assumed to be 210 μC cm−2). The Q value can be calculated from cyclic voltamogram without inclusion of the contribution of the charge from electric double layer. The ECSA calculated for Pt/CNC(700), Pt/CNC(1000), Pt/CNC(1500) and JM catalysts are 76, 67.9, 23.4 and 46.4 m2 g−1, respectively, indicating superior activities of the first two catalysts and thus deserving further studies.
Pt/CNC(700) and JM catalyst was used as the electrocatalysts for a single cell system test. As shown in Supplementary Figure S7, the open cell voltage was about 0.95 V for both Pt/CNC(700) and JM catalysts. At 0.7 V in the activation controlled region, the two catalysts exhibited a similar activity. But below 0.5 V, the current density of the Pt/CNC(700) catalyst was apparently higher than that of JM catalyst at the same voltage. The Pt/CNC(700) catalyst produced a higher maximum power density at a higher current density (0.6 W cm−2, 1.4 A cm−2) than that of JM catalyst (0.55 W cm−2, 1.2 A cm−2).
Thermal stability of Pt/CNC catalyst
The thermal stability test was carried out to examine the stability of CNC supports and Pt particles against air oxidation. The Pt/CNC(700) catalyst was chosen to conduct thermal stability testing because of smaller Pt particle size and less graphitized CNC support, compared with Pt/CNC(1000). Figure 4a shows the XRD patterns of Pt/CNC(700) before and after 2,000 h thermal exposure in air. It is clear that the widths of the platinum peaks do not change significantly during prolonged heating at 80°C. This suggests that the platinum particles did not sinter to form larger grains. Examination of TEM images supports this suggestion. As can be seen in Figure 4b–d, Pt nanoparticles on CNCs dispersed uniformly with no agglomeration or separation from the supports. The particle size of Pt remained about 2.25 nm.

Physical characteristics and electrochemical test of Pt/CNC(700) before and after thermal exposure at 80°C.
(a) XRD patterns, HRTEM images after (b) 500 h, (c) 1,000 h, (d) 2,000 h, (e) cyclic voltammograms of Pt/CNC(700) after thermal exposure and (f) ECSA variation with thermal exposure time.
It is also clear from Figure 4a that the relative signal from the carbon didn't decrease, providing evidence that the support didn't undergo degradation through a combustion process even in the presence of Pt nanoparticles. As a matter of fact, the graphitic structure of CNCs could be still observed after thermal exposure (Figure 4c, d). This is different from the observation made on Pt catalyst supported on an amorphous carbon where carbon signal became little after a long period of thermal exposure in air49.
The chemical composition on the surfaces of Pt/CNC(700) catalysts before and after thermal exposure were analyzed by XPS (Supplementary Figure S8). The regional scan for Pt element provides the valence states of the Pt existing in the catalysts. As shown in Supplementary Figure S8b, c, the Pt signal consists of three pairs of doublets. The most intense doublet (71.2 and 74.6 eV) is due to metallic Pt0. The second set of doublets (71.8 and 75.3 eV) could be assigned to the Pt2+ chemical state as PtO or Pt(OH)2. The third doublet of Pt is the weakest in intensity at even higher binding energies (72.5 and 76.2 eV), which are most likely caused by a small amount of Pt4+ such as PtO2. The percentage of Pt0 can be represented by the relative peak area of the doublet peaks at 71.2 and 74.6 eV. As shown, the content of Pt0 decreased and those of Pt2+ and Pt4+ increased, indicating Pt oxidation during thermal exposure as observed before.
Figure 4e displays the cyclic voltammogram (CV) results of the Pt/CNC(700) catalyst after different hours of thermal exposure at 80°C. In general, the intensity of adsorption/desorption peaks shows a little downtrend from 0 to 500 h. After 500 h, the current density kept unchanged. The ECSA of the catalyst before thermal exposure was about 76 m2 g−1, decreased to about 70 m2 g−1 after 2,000 h thermal exposure as shown in Figure 4f. This observation suggests that Pt/CNC(700) possessed a very high thermal stability. The slight ECSA decrease may result from the decrease of Pt0 and the formation of some oxide on Pt surfaces as revealed by above XPS results.
Electrochemical durability of Pt/CNC catalyst
To investigate the electrochemical durability of the catalyst involved, accelerated degradation testing (ADT) was conducted in a renewable H2SO4 solution (0.5 M) with the catalyst layer exposed to the electrolyte solution to mimic the environment of the electrode membrane interface in PEMFC. Under this specific condition, the deterioration of a catalyst was accelerated. The theoretical open-circuit voltage of PEMFC (zero current) is 1.169 V and for automotive purposes, performances at less than 0.6 V are largely irrelevant6. So in ADT, the voltammetric cycling was first conducted from 0.6 to the extreme limit of 1.24 V, to mimic the operation condition of PEMFC, especially during the time of start-up and shut-down. The cyclic voltammograms at a scan rate of 100 mV s−1 of Pt/CNC(1000) and JM catalysts obtained after potential cycling are shown in Figure 5a, b. The peak currents dropped obviously with potential cycling, indicating a degradation of electrocatalytic activity. The loss of the ECSA of the Pt catalyst with the cycling number is plotted in Figure 5c. After 5,000 cycles, the ECSA decreased from 68 to 34 m2 g−1 for Pt/CNC(1000) (by 50%), from 69 to 13.4 m2 g−1 for Pt/CNC(700) (by 80.6%) and from 46.4 to 7.82 m2 g−1 for JM catalyst (by 83.1%). Therefore, Pt/CNC(1000) showed the best durability. The TEM images and Pt particle size distributions of Pt/CNC(700) and JM catalyst after potential cycling are presented in Supplementary Figures S9 and S10. Pt/CNC(700) illustrated a narrow distribution (3–10 nm) with an average size of 6.35 nm for Pt particles. Quite differently, JM catalyst demonstrated a wider distribution (4–14 nm) with a larger average size of 8 nm. All these is in contrast with the only slight increase of particle size seen with the Pt/CNC(1000) catalyst after equivalent cycling.

Durability test for different catalysts.
Cyclic voltammograms of (a) Pt/CNC(1000) and (b) JM catalyst for various numbers of potential cycling, (c) ECSA variation with cycling number for different catalysts cycled from 0.6 to 1.24 V, cyclic voltammograms of (d) Pt/CNC(1000), (e) JM catalyst for various numbers of potential cycling and (f) ECSA variation with cycling number for different catalysts cycled from 0.6 to 1.0 V.
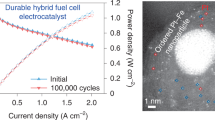
In practical operation, measured open-circuit voltages are usually below 1.0 V, being lower than theoretical ones owing to imperfect separation of the gases by the membrane and its finite electronic resistance. So the potential cycling was further carried out in a potential range from 0.6 to 1.0 V at a scan rate of 50 mV s−1, to mimic the practical operation condition of PEMFC. Figure 5d, e represents the changes of voltammograms during ADT at different cycles and the ECSA variation with cycling is shown in Figure 5f. As can be seen, the peak current for JM catalyst decreased as cycling proceeded. The ECSA dropped from 51 to 33.5 m2 g−1 and about 65.6% of its initial ECSA remained. However, Pt/CNC(1000) showed a slight increase in ECSA in the first 1,000 cycles. From 1,000 to 4,000 cycles, the ECSA decreased a little, but was still higher than its original value. In order to confirm this high stability, 2,000 more cycles were conducted. It was found that the ECSA showed a small fluctuation (from 57 to 62 m2 g−1) and stayed at the level of about 110% of its initial value. These results provide evidence for the superior durability of Pt/CNC(1000) catalyst with extended long potential cycling over that for JM catalyst.
The polarization curves for the ORR obtained with different catalysts on a rotating disk electrode (RDE) in O2-saturated 0.5 M H2SO4 solution at 1,600 rpm before and after ADT are shown in Figure 6. For the initial test, all electrodes were pre-treated by cycling the potential between 0 to 1.24 V at a sweep rate of 100 mV s−1 for 10 cycles to remove surface contamination. It can be seen in Figure 6 that all of the catalysts had a similar onset potential of about 900 mV. In polarization curves, the half-wave potential (the potential corresponding to one-half of the diffusion current), E1/2, was used to evaluate the ORR electrochemical activity of a catalyst. Pt/CNC(1000) and Pt/CNC(700) showed an E1/2 of about 804 mV and 800 mV, respectively. But, the E1/2 for JM catalyst was 762 mV, about 40 mV left shift compared with Pt/CNCs.

ORR polarization curves of different catalysts.
(a) Pt/CNC(1000), (b) Pt/CNC(700), (c) JM catalyst before and after potential cycling between 0.6 to 1.24 V (1,600 rpm, scan rate: 5 mV s−1) and (d) ORR polarization curves for Pt/CNC(1000) before and after potential cycling between 0.6 to 1.0 V (1,600 rpm, scan rate: 5 mV s−1).
All the catalysts showed degradation in half-wave potential within the potential cycling from 0.6 to 1.24 V at a scan rate of 100 mV s−1. As shown in Figure 6a, the E1/2 of Pt/CNC(1000) shifted to 727 mV, with a decrease of 77 mV after 1,000 cycles. And after 3,000 cycles, the Pt/CNC(1000) showed a degradation of 169 mV in its half-wave potential (635 mV). However, for Pt/CNC(700), although the initial E1/2 was similar to that of Pt/CNC(1000), after 1,000 cycles, the E1/2 shifted to 657 mV with a deterioration of 143 mV. Moreover, the degradation after 3,000 cycles was much larger, with a 340 mV negative shift in E1/2 (460 mV). These results confirm that supports played a crucial role for the durability of Pt catalyst. For comparison, the ORR polarization curves of JM catalyst are shown in Figure 6c. After 1,000 cycles, the E1/2 of JM catalyst shifted to 654 mV, with a 108 mV degradation. And after 2,000 cycles, JM catalyst showed a degradation of 190 mV in half-wave potential E1/2, indicating a lower durability than Pt/CNC(1000). The difference in half-wave potential between the Pt/CNC(1000) and Pt/CNC(700) catalysts increased to 175 mV after 3,000 cycles. Furthering cycling revealed that the difference increased to higher values after more cycles.
Figure 6d shows the ORR polarization curves for Pt/CNC(1000) before and after potential cycling between the practical range of 0.6 to 1.0 V at a scan rate of 50 mV s−1. The curves nearly coincided. These experiments were repeated a few times and consistent results were observed. For comparison, other catalysts were also tested in the range of 0.6 to 1.0 V, which generally showed activity degradation, but at very slow rates. For example, a degradation of several tens of mV in E1/2 was observed after 4,000 potential cycles for Pt/CNC(700) (Supplementary Figure S11). This degradation rate is much slower than that observed in the range of 0.6 to 1.24 V (Fig. 6b). Such results indicate that potential cycling in the range of 0.6–1.0 V may not be suitable for ADT because the activity degradation is slow and the performance may not be differentiable for different catalysts within several thousands of cycling.
Discussion
The present experiments based on both half and single cells clearly show that the catalysts of Pt/CNC(700) and Pt/CNC(1000) have higher catalytic activities toward HOR and ORR than JM catalyst (Figures 5a and 6a–c, Figures S6b and S7). Furthermore, these catalysts, particularly Pt/CNC(1000), exhibits remarkably higher long-term stabilities for both thermal oxidation (Figure 4f) and electrochemical corrosion (Figures 5c and 6a). When tested under conditions of practical operation of PEMFC, Pt/CNC(1000) appeared extremely stable and indicated almost no degradation during the long-term testing for HOR and ORR (Figures 5f and 6d). ORR at cathode is several orders of magnitude slower than HOR at anode and thus rate-controlling for the overall chemical reaction in fuel cell6. Any small enhancement in ORR activity could lead to apparent improvement in the overall performance. Considering the high activity, especially the drastically high durability for ORR, the present catalyst of Pt/CNC(1000) represents a significant advance over the Pt catalyst currently in wide use. Incorporation of this new catalyst could hopefully extend the current lifetime of 2,000–3,000 h of PEMFC to the targeted 5,000 h. This, surely, needs to be verified by extensive on-road testing.
The significant improvement in catalytic performance can be attributed to the unique features of the support material of CNCs. The present CNCs are spherical and hollow and thus have a high surface area. Particularly, they can be well dispersed in chemical solutions for uniform Pt deposition and uniform catalyst ink preparation after Pt deposition. This is quite different from previous observations made from the use of CNTs29,30. CNTs are generally long and entangled and thus difficult to be dispersed in solutions for catalyst and MEA preparations. Cutting to short lengths and surface functionalization are approaches having been practiced for dispersion improvement, but these approaches usually induce structure and property degradations to CNTs.
The further reason for the significant improvement in catalytic performance can be that the CNCs have a graphitic structure with N-doping. The immediate advantage of N doping can be clearly seen from the results of Pt deposition and catalytic activity. As shown in Figure 3, Pt nanoparticles dispersed on the CNCs prepared at 700°C uniformly with small and narrowly distributed particle size. However, with enhancing graphitization degree, the particle size increased and agglomeration took place, especially in the sample of Pt/CNC(1500).
It is well known that support chemistry and support/catalyst interactions play a significant role in determining Pt nanoparticle size and dispersion since the nature of the support governs the nucleation and growth processes during Pt deposition. The CNCs prepared at a high temperature of 1,500°C had a well-developed graphitic structure but with fewer defect locations for the nucleation of Pt, consequently a larger particle size results. The CNCs prepared at temperatures of 1,000 and 700°C had not only good graphitic structures but some amount of doped N as well. The incorporation of nitrogen atoms into carbon supports is believed to provide enhanced nucleation of catalyst particles as C–N defects and N interstitials can serve as local heterogeneous Pt nucleation sites50,51,52,53,54. This may be the origin of uniform dispersion of fine Pt particles on graphitic supports in Pt/CNC(700) and Pt/CNC(1000). Compared with these catalysts, the Pt dispersion in JM catalyst (Supplementary Figure S10a, b) using an amorphous carbon support was not uniform. This observation again confirms the beneficial effect of nitrogen doping. Since the electrochemical activity is closely associated with the size and dispersion of Pt particles, Pt/CNC(700) and Pt/CNC(1000), having fine and well dispersed Pt particles, demonstrated a better activity than Pt/CNC(1500) and JM catalyst.
It should be noted that N doping in conventional CNTs is usually above 3% for bringing about good deposition of Pt on them. However, the present CNCs contained lower contents of N (~2.20% and 0.96% for the 700°C and 1000°C samples, respectively) and excellent dispersion of Pt nanoparticles were clearly observed. This might be owing to the fact that the graphitic structure of CNTs is made up of only hexagonal C rings, but the structure of CNCs consists of not only such rings but pentagonal and other C rings as well. That is, CNCs contain more favorable structural sites for Pt deposition than CNTs.
Catalyst durability is primarily determined by the resistance of support material to corrosion. Enhancing graphitization is beneficial to durability improvement. Nitrogen doping is known to improve the stability of carbon materials. But, opposite results also exist in the literature. It was found that high contents of nitrogen would decrease the stability of carbon materials, because of the structural disorder introduced by the presence of N into the carbon lattice. Moreover, with increasing nitrogen content, the oxidation resistance of mesoporous carbon could decrease because excessive nitrogen doping might accelerate carbon oxidative gasfication55. In the potential cycling test, the corrosion of carbon and consequent detachment of Pt particles accelerated catalytic degradation. Comparing different supports, Pt/CNC(1000) exhibited the best electrochemical durability in ADT. Especially in ORR, the difference of E1/2 between Pt/CNC(700) and Pt/CNC(1000) after 3,000 cycles was even as large as 175 mV. This is a result of the good graphitic structure of CNCs prepared at 1,000°C, which is resistant to electrochemical corrosion. Moreover, in the potential cycling between 0.6 to 1.0 V, there was almost no degeneration in both HOR and ORR for Pt/CNC(1000). Such superior stability could be a consequence of the graphitic CNC support that could be little corroded within this potential range of the practical operation of PEMFC.
Catalyst durability is also determined by the interaction of support with Pt particles. It is believed that increasing the graphitization degree of carbon support leads to the enhanced π sites on carbon and these sites act as anchoring centers for Pt nanoparticles56,57. Thus, graphitization to a higher degree strengthens the interaction between Pt particles and the carbon support. This is supported by results on the binding energies of Pt 4f7/2 on core-level XPS spectra for the catalysts of Pt/VXC, Pt/CNC (undoped) and Pt/CNC(700) (Supplementary Figure S12). The strengthening of Pt-support interaction could lead to a high resistance to the surface migration of Pt particles. As a result, the Pt catalyst such as Pt/CNC(1000) using highly graphitic CNCs showed the best electrochemical durability.
The Pt-support interaction can be further strengthened with N doping into the graphitic structure. Nitrogen functionalities act as “tethers” (chemical sites binding with Pt particles), increasing catalytic activity not only by improving dispersion of Pt particles but also by providing strong binding and thus resistance to nanoparticle agglomeration and coarsening51,58,59. The advantage of N doping can be seen from the results of thermal stability. Pt nanoparticles on CNCs dispersed uniformly with no agglomeration or separation from the supports even after 2,000 hours of thermal exposure in air, leading to almost no ECSA loss for Pt/CNC(700) (Figure 4). This differs from our observation made on a Pt catalyst also supported on graphitic CNCs, but without N doping. In the latter case, the Pt nanoparticles grew bigger with an apparent ECSA degeneration after 1,000 h thermal exposure.
Nitrogen atom has one more electron than carbon atom, which alters the electron donating character of the support, which in turn impacts the binding of the catalyst to the support. At locations of carbon already substituted by nitrogen, the interaction between Pt nanoparticles and the delocalized π bond is enhanced60. Moreover, pyridinic N species occupy edge plane sites and thereby carry an extra lone pair of electrons. Such edge sites may provide stronger Pt-support interaction, thus inhibiting agglomeration of Pt nanoparticles.
It is still difficult to identify the specific functionalities responsible for the improved stability due to the complex and heterogeneous nature and overlap of the binding energies of involved N species. The change of the binding energy of Pt 4f7/2 (Supplementary Figure S12) indicates an intra-atomic charge transfer from the support to the Pt atom, resulting in strengthening of the interaction between supports and Pt nanoparticles. Therefore, it is graphitization and N doping that strengthened the Pt-support interaction and thus enhanced the thermal and electrochemical stabilities observed for Pt/CNC(700) and Pt/CNC(1000).
The dissolution and redeposition of Pt particles are also a mechanism of catalyst degradation. The dissolution of Pt particles is also affected by their interaction with carbon support. Strengthened interaction with N-doped graphitic CNCs, as discussed above, could increase electron donation from support to the surface of Pt and the minimum potential required to electrochemically oxidize Pt. Meanwhile, N doping promotes uniform redeposition of Pt on carbon support. As shown in Supplementary Figures S9 and S10, Pt/CNC(700) illustrated a more uniform distribution of a smaller Pt size than JM catalyst after the same 5,000 cycles. This evidences, again, the beneficial effects of N doping and graphitic structuring for stabilizing Pt particles and thus catalytic activity.
In summary, graphitic hollow CNCs were prepared by spray pyrolysis and heat treatment with sulfur. In situ N doping was achieved by using pyridine as C and N precursors. Such material was used as the support for Pt particles and the catalytic performance of the resultant catalyst of Pt/CNC was evaluated from half and single cell testing. N doping was found to be essential to having uniform deposition of fine Pt nanoparticles on graphitic CNC support and improved catalytic activity toward HOR and ORR. Results of ADT elucidated that both thermal stability and electrochemical durability were drastically enhanced for Pt/CNC(1000) with N doping and highly graphitic CNCs, when compared with Pt/CNC(700) with also N doping but less graphitized CNCs and JM catalyst with no N doping and little graphitized carbon black. When tested under potential conditions of practical fuel cell operation, Pt/CNC(1000) exhibited almost no degradation for both HOR and ORR, when compared with the apparent degradation observed for JM catalyst. Such a remarkable catalytic activity and durability were attributed to the excellent comprehensive properties of CNCs required for a catalyst support, particularly their strong oxidation resistance and strong interaction with Pt particles due to their graphitization and concurrent N doping. The superior performance of the present catalyst over JM catalyst of an industry benchmark sheds light on how to overcome the shortcoming of current catalyst technology and extend the lifetime to reach the durability target. Since the present CNCs and Pt catalyst have many unique physical and chemical properties, they can be used not only in PEMFC but also in many other areas such as fuel reforming, hydrogen production and industrial chemical applications.
Methods
Preparation of hollow N-doped CNCs
The hollow CNCs were prepared by a spray pyrolysis method. Firstly, iron/graphite core-shell nanoparticles were obtained by thermal decomposition the mixture of pyridine (C5H5N) and pentacarbonyl [Fe(CO)5] at 700 and 1,000°C. Pyridine (C5H5N) was used as carbon and nitrogen sources. For comparison, such particles were also prepared at 1,500°C, but acetylene (C2H2) was used as carbon source in this case. Secondly, the Fe core was removed by heat treatment with inclusion of sulfur. To do so, the original samples were mixed with sulfur powder and underwent heat treatment at 900°C. Then, the heated sample was mixed in dilute HCl (HCl:H2O = 1:1) and filtered using distilled water. The sample collected on the filter was dried at 100°C for 2 h.
Preparation of Pt catalyst supported on CNCs
The Pt catalyst supported on CNCs was prepared by an ethylene glycol (EG) reduction method. Typically, carbon supports were sonicated in EG first. Then, a chloroplatinic acid (CPA) solution, prepared by dissolving CPA in EG, was added into the carbon slurry and then stirred for 20 minutes. The nominal amount of Pt deposited was set to be 45 wt.% (weight percentage). The mixture was heated up to 130°C and refluxed for 3 h under continuous magnetic stirring. After having been cooled down to room temperature, the catalyst was filtered and washed with deionized water several times until no Cl− could be detected by AgNO3 solution. The CNC supports used were originally prepared at 700, 1,000, 1,500°C and the catalysts are designed as Pt/CNC(700), Pt/CNC(1000) and Pt/CNC(1500), respectively.
Characterization of CNCs and Pt/CNC
X-ray diffraction (XRD) was operated at 35 kV and 200 mA with Nickel-filtered Cu-Kα radiation as incident beam (D/max 2550VL/PC), to study the crystallization of carbon and other phases contained in the samples. Typical transmission electron microscopy (TEM) and high resolution transmission electron microscopy (HRTEM) with a cold field emission gun (JEOL-2010F) were used to investigate the microstructure and morphology of the samples. Energy-dispersive X-ray spectroscopy (EDS, Oxford INCA 200 attached to the HRTEM instrument) was also employed to analyze elemental compositions. Raman spectroscopy was used to examine the perfection of the hollow CNCs using a Horiba Yvon HR 800UV with a 514.5 nm excitation wavelength laser. Thermogravimetric analysis (TGA) (TGA/SDTA/DSC851e) was conducted by heating samples to 900°C at 10°C min−1 in an air atmosphere. The surface of the CNCs was analyzed with Kratos Axis Ultra DLD X-ray photoelectron spectroscopy (XPS). This system uses a focused monochromated Al Kα (15 kV) X-ray source for excitation and a spherical section analyzer at a base pressure of 10−9 Torr. The peak energies were calibrated by placing the major C1s peak at 284.8 eV.
Electrochemical testing
Since catalyst stability involves thermal stability and electrochemical durability, we examined them separately. For the study of thermal stability, the Pt/CNC(700) catalyst was placed in an oven with dry air at 80°C for up to 2,000 h. At a certain interval of time, a small amount of the sample was taken out for electrochemical analysis by using a cyclic voltammetry technique. The electrochemical experiments were performed in a three-electrode cell using a CHI618D instrument at a constant temperature. This temperature was controlled to be within 25 ± 2°C in a water bath in order to minimize the effect of ambient temperature variation from winter to summer or from day to night time. The reference electrode and counter electrode were saturated calomel electrode (SCE) and large-area Pt plate, respectively and 0.5 M H2SO4 was used as the electrolyte.
The working electrode was prepared as follows: 17.5 mg catalyst was dispersed in a mixture of 4.65 ml ethanol and 0.35 ml 5 wt.% Nafion solution. The mixture was agitated by ultrasonication for 30 min to form an ink. A fixed amount of this ink was then dropped on a glassy carbon electrode with a diameter of 3 mm and dried to yield a thin-film electrode. The metal loading on the electrode was 0.2 or 0.4 mg cm−2. All potentials in this work were referred to reversible hydrogen electrode (RHE). Before electrochemical test, the electrolyte was purged with N2 or O2 for more than 30 minutes and the gas purging continued during the test to maintain the gas saturated condition. The cyclic voltammetry was conducted over a potential range from 0.6 to 1.24 V at a scan rate of 100 mV s−1, or from 0.6 to 1.0 V at a scan rate of 50 mV s−1 for several thousand cycles in accelerated durability testing (ADT). At a certain interval of cycles, electrochemical test was conducted in a renewable gas saturated electrolyte.
The catalytic activity for oxygen reduction reaction (ORR) was evaluated using a rotating disk electrode (RDE, Pine, AFMSRCE 3005) by linear sweep voltammetry (LSV). The working electrode was prepared as follows: 4 mg catalyst was dispersed in a mixture of methanol and 5 wt.% Nafion solution with a weight ratio of 50:1. The mixture was agitated by ultrasonication for 30 min to form an ink. A given amount of this ink was then dropped on a glassy carbon electrode with a diameter of 5.6 mm and dried to yield a thin-film electrode. The Pt loading on the RDE was controlled to be 34 μg cm−2. The ORR polarization curves were obtained in O2 saturated 0.5 M H2SO4 at 25 ± 2°C with a scan rate of 5 mV s−1 and a rotation speed of 1,600 rpm.
For comparison, a Pt catalyst supported on Vulcan XC-72R carbon black with 40 wt.% Pt loading from Johnson Matthey Co. (JM) was also deposited on electrodes and tested with the same procedures. The Pt loading of this catalyst was controlled to be the same as that for Pt/CNC catalysts. This was to provide a baseline state-of-the-art commercially available catalyst, so that improvements can be easily identified from current catalyst technologies.
References
Service, R. F. Shrinking fuel cells promise power in your pocket. Science 296, 1222–1224 (2002).
Herring, A. M., Zawodzinski Jr, T. A. & Hamrock, S. J. Fuel Cell Chemistry and Operation [1–13] (American Chemical Society, Washington DC, 2010).
Vogel, J. FC40 international stationary fuel cell demonstration. In: DOE Hydrogen Program 2008 Annual Progress Report V. Fuel Cells, 912–915 (2008). Date of access: 05/11/2008, at <http://www.hydrogen.energy.gov/pdfs/progress08/v_d_4_vogel.pdf>.
Milliken, J. Hydrogen, fuel cells & infrastructure technologies program. Multi-Year Research, Development and Demonstration Plan. Planned program activities for 2005–2015. Page 3.4–14 (2007). Date of access: 01/10/2007 at <http://www.nrel.gov/docs/fy08osti/39146.pdf>.
Gasteiger, H. A. & Marković, N. M. Just a dream–or future reality? Science 324, 48–49 (2009).
Debe, M. K. Electrocatalyst approaches and challenges for automotive fuel cells. Nature 486, 43–51 (2012).
Wang, Y. J., Wilkinson, D. P. & Zhang, J. Noncarbon support materials for polymer electrolyte membrane fuel cell electrocatalysts. Chem. Rev. 111, 7625–7651 (2011).
Borup, R. et al. Scientific aspects of polymer electrolyte fuel cell durability and degradation. Chem. Rev. 107, 3904–3951 (2007).
Tang, H., Qi, Z., Ramani, M. & Elter, J. F. PEM fuel cell cathode carbon corrosion due to the formation of air/fuel boundary at the anode. J. Power Sources 158, 1306–1312 (2006).
Kadirgan, F. et al. Carbon supported nano-sized Pt–Pd and Pt–Co electrocatalysts for proton exchange membrane fuel cells. Int. J. Hydrogen Energy 34, 9450–9460 (2009).
Mathias, M. et al. Two fuel cell cars in every garage? Interface 14, 24–35 (2005).
Cameron, D. S. Fuel cells science and technology 2008. Platinum Met. Rev. 53, 147–154 (2009).
Franco, A. A., Guinard, M., Barthe, B. & Lemaire, O. Impact of carbon monoxide on PEFC catalyst carbon support degradation under current-cycled operating conditions. Electrochim. Acta 54, 5267–5279 (2009).
Büchi, F., Inaba, M. & Schmidt, T. Polymer Electrolyte Fuel Cell Durability. [29–53] (Springer, 2009).
Knights, S. D., Colbow, K. M., St-Pierre, J. & Wilkinson, D. P. Aging mechanisms and lifetime of PEFC and DMFC. J. Power Sources 127, 127–134 (2004).
Wipke, K. et al. Controlled hydrogen fleet and infrastructure analysis. In: 2011 DOE Hydrogen Program Annual Merit Review and Peer Evaluation Meeting. Washington, DC (2011). Date of access: 13/05/2011 at <http://www.hydrogen.energy.gov/pdfs/review11/tv001_wipke_2011_o.pdf>.
Shao, Y., Liu, J., Wang, Y. & Lin, Y. Novel catalyst support materials for PEM fuel cells: current status and future prospects. J. Mater. Chem. 19, 46–59 (2009).
Zhou, J. et al. Interaction between Pt nanoparticles and carbon nanotubes – An X-ray absorption near edge structures (XANES) study. Chem. Phys. Lett. 437, 229–232 (2007).
Si, Y. & Samulski, E. T. Exfoliated graphene separated by platinum nanoparticles. Chem. Mater. 20, 6792–6797 (2008).
Zhang, S. et al. Graphene decorated with PtAu alloy manoparticles: facile synthesis and promising application for formic acid oxidation. Chem. Mater. 23, 1079–1081 (2011).
Li, Y., Gao, W., Ci, L., Wang, C. & Ajayan, P. M. Catalytic performance of Pt nanoparticles on reduced graphene oxide for methanol electro-oxidation. Carbon 48, 1124–1130 (2010).
Deng, D. et al. Toward N-doped graphene via solvothermal synthesis. Chem. Mater. 23, 1188–1193 (2011).
Lee, J., Kim, J. & Hyeon, T. Recent progress in the synthesis of porous carbon materials. Adv. Mater. 18, 2073–2094 (2006).
Gan, L., Lv, R., Du, H., Li, B. & Kang, F. Highly dispersed Pt nanoparticles by pentagon defects introduced in bamboo-shaped carbon nanotube support and their enhanced catalytic activity on methanol oxidation. Carbon 47, 1833–1840 (2009).
Li, S. et al. Green synthesis of a Pt nanoparticle/polyoxometalate/carbon nanotube tri-component hybrid and its activity in the electrocatalysis of methanol oxidation. Carbon 49, 1906–1911 (2011).
Chang, Y., Han, G., Li, M. & Gao, F. Graphene-modified carbon fiber mats used to improve the activity and stability of Pt catalyst for methanol electrochemical oxidation. Carbon 49, 5158–5165 (2011).
Shao, Y., Yin, G., Gao, Y. & Shi, P. Durability study of Pt/C and Pt/CNTs catalysts under simulated PEM fuel cell conditions. J. Electrochem. Soc. 153, A1093–A1097 (2006).
Wang, X., Li, W., Chen, Z., Waje, M. & Yan, Y. Durability investigation of carbon nanotube as catalyst support for proton exchange membrane fuel cell. J. Power Sources 158, 154–159 (2006).
Wang, X. X. & Wang, J. N. Preparation of short and water-dispersible carbon nanotubes by solid-state cutting. Carbon 46, 117–125 (2008).
Wang, X. X., Wang, J. N., Chang, H. & Zhang, Y. F. Preparation of short carbon nanotubes and application as an electrode material in Li-Ion batteries. Adv. Funct. Mater. 17, 3613–3618 (2007).
Panizza, M. & Cerisola, G. Application of diamond electrodes to electrochemical processes. Electrochim. Acta 51, 191–199 (2005).
Wang, J. & Swain, G. M. Dimensionally stable Pt/diamond composite electrodes in concentrated H3PO4 at high temperature. Electrochem. Solid-State Lett. 5, E4–E7 (2002).
Gao, J. S. et al. Preparation and characterization of metal nanoparticles on a diamond surface. Chem. Mater. 12, 3495–3500 (2000).
Fischer, A. E. & Swain, G. M. Preparation and characterization of boron-doped diamond powder: a possible dimensionally stable electrocatalyst support material. J. Electrochem. Soc. 152, B369–B375 (2005).
Zang, J. B., Wang, Y. H., Huang, H. & Tang, W. Electrochemical behavior of high-pressure synthetic boron doped diamond powder electrodes. Electrochim. Acta 52, 4398–4402 (2007).
Su, F. et al. Preparation and characterization of highly ordered graphitic mesoporous carbon as a Pt catalyst support for direct methanol fuel cells. Chem. Mater. 17, 3960–3967 (2005).
Choi, W. C. et al. Platinum nanoclusters studded in the microporous nanowalls of ordered mesoporous carbon. Adv. Mater. 17, 446–451 (2005).
Chai, G. S., Yoon, S. B., Yu, J. S., Choi, J. H. & Sung, Y. E. Ordered porous carbons with tunable pore sizes as catalyst supports in direct methanol fuel cell. J. Phys. Chem. B 108, 7074–7079 (2004).
Joo, S. H. et al. Ordered mesoporous carbons (OMC) as supports of electrocatalysts for direct methanol fuel cells (DMFC): effect of carbon precursors of OMC on DMFC performances. Electrochim. Acta 52, 1618–1626 (2006).
Joo, J. B., Kim, P., Kim, W., Kim, J. & Yi, J. Preparation of mesoporous carbon templated by silica particles for use as a catalyst support in polymer electrolyte membrane fuel cells. Catal. Today 111, 171–175 (2006).
Nakagawa, N., Suzuki, Y., Watanabe, T., Takei, T. & Kanamura, K. Preparation of Pt-Ru nanoparticles with a uniform size distribution on a mesoporous carbon and their activity towards methanol electro-oxidation. Electrochemistry 75, 172–174 (2007).
Chan, K. Y., Ding, J., Ren, J., Cheng, S. & Tsang, K. Y. Supported mixed metal nanoparticles as electrocatalysts in low temperature fuel cells. J. Mater. Chem. 14, 505–516 (2004).
Fuertes, A. B. & Alvarez, S. Graphitic mesoporous carbons synthesised through mesostructured silica templates. Carbon 42, 3049–3055 (2004).
Sevilla, M. & Fuertes, A. B. Catalytic graphitization of templated mesoporous carbons. Carbon 44, 468–474 (2006).
Tuinstra, F. & Koenig, J. L. Raman spectrum of graphite. J. Chem. Phys. 53, 1126–1130 (1970).
Nemanich, R. J. & Solin, S. A. First- and second-order Raman scattering from finite-size crystals of graphite. Phys. Rev. B 20, 392–401 (1979).
Antolini, E., Giorgi, L., Pozio, A. & Passalacqua, E. Influence of Nafion loading in the catalyst layer of gas-diffusion electrodes for PEFC. J. Power Sources 77, 136–142 (1999).
Lee, E. P. et al. Growing Pt nanowires as a densely packed array on metal gauze. J. Am. Chem. Soc. 129, 10634–10635 (2007).
Stevens, D. A. & Dahn, J. R. Thermal degradation of the support in carbon-supported platinum electrocatalysts for PEM fuel cells. Carbon 43, 179–188 (2005).
Zhou, Y. et al. Enhancement of Pt and Pt-alloy fuel cell catalyst activity and durability via nitrogen-modified carbon supports. Energy Environ. Sci. 3, 1437–1446 (2010).
Zhou, Y. et al. Improving PEM fuel cell catalyst activity and durability using nitrogen-doped carbon supports: observations from model Pt/HOPG systems. J. Mater. Chem. 19, 7830–7838 (2009).
Zhou, Y. et al. Dopant-induced electronic structure modification of HOPG surfaces: implications for high activity fuel cell catalysts. J. Phys. Chem. C 114, 506–515 (2009).
Brian, H. M. & Alberto, S. Platinum nanoparticles on carbonaceous materials: the effect of support geometry on nanoparticle mobility, morphology and melting. Nanotechnology 19, 195711 (2008).
Wiggins-Camacho, J. D. & Stevenson, K. J. Effect of nitrogen concentration on capacitance, density of states, electronic conductivity and morphology of N-doped carbon nanotube electrodes. J. Phys. Chem. C 113, 19082–19090 (2009).
Nxumalo, E. N. & Coville, N. J. Nitrogen doped carbon nanotubes from organometallic compounds: a review. Materials 3, 2141–2171 (2010).
Shao, Y., Yin, G. & Gao, Y. Understanding and approaches for the durability issues of Pt-based catalysts for PEM fuel cell. J. Power Sources 171, 558–566 (2007).
Wang, S., Jiang, S. P., White, T. J., Guo, J. & Wang, X. Electrocatalytic activity and interconnectivity of Pt nanoparticles on multiwalled carbon nanotubes for fuel cells. J. Phys. Chem. C 113, 18935–18945 (2009).
Ye, S., Vijh, A. K. & Dao, L. H. A new fuel cell electrocatalyst based on carbonized polyacrylonitrile foam: the nature of platinum-support interactions. J. Electrochem. Soc. 144, 90–95 (1997).
Maiyalagan, T., Viswanathan, B. & Varadaraju, U. V. Nitrogen containing carbon nanotubes as supports for Pt-Alternate anodes for fuel cell applications. Electrochem. Commun. 7, 905–912 (2005).
Coloma, F., Sepulveda-Escribano, A., Fierro, J. L. G. & Rodriguez-Reinoso, F. Preparation of platinum supported on pregraphitized carbon blacks. Langmuir 10, 750–755 (1994).
Acknowledgements
Financial supports from National Natural Science Foundation of China (project #: 51271077, U1362104) and Shanghai Nanoscience and Nanotechnology Promotion Center (project #: 12 nm0503300) are greatly acknowledged.
Author information
Authors and Affiliations
Contributions
J.N.W. designed the research program. X.X.W., Z.H.T. and M.Z. carried out the experiments. X.X.W. and J.N.W. analyzed the data and wrote the manuscript. All authors reviewed, discussed and approved the results and conclusions of this Article.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Wang, X., Tan, Z., Zeng, M. et al. Carbon nanocages: A new support material for Pt catalyst with remarkably high durability. Sci Rep 4, 4437 (2014). https://doi.org/10.1038/srep04437
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep04437
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
