
Abstract
Blood-brain barrier (BBB) models are often used to investigate BBB function and screen brain-penetrating therapeutics, but it has been difficult to construct a human model that possesses an optimal BBB phenotype and is readily scalable. To address this challenge, we developed a human in vitro BBB model comprising brain microvascular endothelial cells (BMECs), pericytes, astrocytes and neurons derived from renewable cell sources. First, retinoic acid (RA) was used to substantially enhance BBB phenotypes in human pluripotent stem cell (hPSC)-derived BMECs, particularly through adherens junction, tight junction and multidrug resistance protein regulation. RA-treated hPSC-derived BMECs were subsequently co-cultured with primary human brain pericytes and human astrocytes and neurons derived from human neural progenitor cells (NPCs) to yield a fully human BBB model that possessed significant tightness as measured by transendothelial electrical resistance (~5,000 Ωxcm2). Overall, this scalable human BBB model may enable a wide range of neuroscience studies.
Similar content being viewed by others
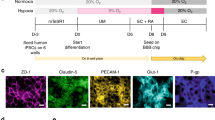


Introduction
The blood-brain barrier (BBB) is composed of brain microvascular endothelial cells (BMECs) which line brain capillaries and control molecular and cellular trafficking between the bloodstream and neural tissue. These properties are tightly regulated by the surrounding neurovascular microenvironment throughout BBB development and into adulthood. While this barrier is essential for preserving healthy brain activity, its dysfunction has been implicated in a number of neurological diseases1. Moreover, an intact BBB serves as a major bottleneck for brain drug delivery2. Studies regarding BBB development and regulation can be difficult and time-consuming to conduct in vivo and testing brain penetration of therapeutics in vivo is a low throughput endeavor. As such, in vitro BBB models have been widely implemented to study interactions between BMECs and other cells of the neurovascular unit and to conduct screens for prospective BBB-permeant drugs.
In vitro BBB models are typically constructed using primary BMECs isolated from animal brain tissue, including bovine, porcine, rat and mouse (reviewed extensively in ref. 3). These BMECs are then co-cultured with combinations of other cells of the neurovascular unit, such as neurons, pericytes and astrocytes, to upregulate BBB properties4,5,6,7. Models derived from animal tissue have proven extremely useful in studying various aspects of the BBB, such as developmental and regulatory mechanisms8,9,10,11,12 and assaying drug permeability, but it is generally well-accepted that owing to species differences, a robust human BBB model is vital to achieve a detailed understanding of human developmental pathways and to conduct relevant drug discovery and design studies13. Human BMEC sources for BBB models have previously consisted of either primary biopsied brain tissue14,15 or immortalized cell lines16. Primary human BMECs typically possess moderate barrier properties but are of limited scale14,15 and immortalized BMECs are clonal and readily scalable but often suffer from suboptimal barrier properties16,17. From a co-culture perspective, human neurons, astrocytes and pericytes can also be difficult to obtain from primary tissue sources in sufficient quantities for modeling purposes. These collective issues have hindered the development of in vitro human BBB models that are both high fidelity and scalable3.
We have recently demonstrated that stem cells may be attractive candidates to replace primary cells in human BBB models. Human pluripotent stem cells (hPSCs), including both human embryonic stem cells (hESCs) and induced pluripotent stem cells (iPSCs), can be differentiated into cells possessing both endothelial and BBB properties (coined hPSC-derived BMECs) via co-differentiation of neural and endothelial progenitors, followed by selection and subsequent culture of the endothelial cells18. The iPSC-derived BMECs co-cultured with rat astrocytes possessed reasonable barrier tightness as measured by TEER (860 ± 260 Ωxcm2)18, but the TEER remained below some primary bovine19 and porcine20,21 models (800–2,000 Ωxcm2) and substantially lower than in vivo TEER (measured up to 5,900 Ωxcm2)22. In searching for candidates to improve the BBB phenotype, we identified all-trans retinoic acid (RA). BMECs in vivo have been shown to express retinol-binding protein and its membrane receptor STRA623, STRA6 expression has been detected in brain endothelium but not peripheral endothelium in adult mice24 and STRA6 expression was increased during the differentiation of hPSC-derived BMECs in our previous work18. Moreover, RA has been shown to upregulate certain BBB properties in immortalized rodent25,26 and human27 BMEC lines. In this manuscript, we demonstrate maturation of hPSC-derived BMEC phenotypes following retinoic acid (RA) addition during the differentiation process, including enhanced adherens junction protein expression, barrier function and multidrug resistance protein (MRP) efflux activity. We also demonstrated in previous work that primary human neural progenitor cells (NPCs) could be differentiated to a defined mixture of neurons and astrocytes capable of inducing BBB properties in rat BMECs in co-culture7. In this manuscript, it is shown that under optimized culture conditions, RA-treated hPSC-derived BMECs sequentially co-cultured with primary human brain pericytes and NPC-derived astrocytes and neurons can achieve physiologic TEER values, forming a scalable, fully human BBB model.
Results
RA enhances BBB properties in hPSC-derived BMECs
IMR90-4 iPSCs were first differentiated for 6 days in unconditioned medium (UM) to generate a mixed population of neural cells and immature BMECs as previously described18. Then, RA was added during the 2 days of EC medium treatment (Fig. 1A) and resultant cells were analyzed. RA-treated cultures exhibited similar profiles of vascular and BBB markers as described previously18, with PECAM-1, GLUT-1, claudin-5 and occludin all widely expressed in the differentiating cell population (Fig. 1B). However, whereas cultures under the standard EC treatment protocol did not express VE-cadherin until after passaging and purification on collagen/fibronectin matrix18, RA treatment during the EC treatment phase substantially induced VE-cadherin expression prior to passage and purification and the effect was dose dependent requiring 10 μM RA (Fig. 1B–D). After 2 days of RA treatment in EC medium, 27 ± 6% of the cultures expressed VE-cadherin (Fig. 1C). By comparison, the epithelial marker E-cadherin, which is expressed in undifferentiated hPSCs28, was not detected by flow cytometry and exhibited weak junctional expression by immunocytochemistry in both control and RA-treated cells after the EC treatment phase (Supplementary Fig. S1 online). In addition to modulating adherens junctions by increased VE-cadherin expression, RA treatment also led to changes in tight junction protein expression yielding a 4-fold increase in occludin expression and a 1.7-fold decrease in claudin-5 expression (Fig. 1D).

RA treatment modulates BBB properties in differentiating IMR90-4 iPSCs.
10 μM RA was used for all experiments except those in panel D. (A) All data were collected from mixed, differentiating cultures after 48 h of EC medium treatment with or without RA. (B) Expression of PECAM-1, GLUT-1, occludin, claudin-5 and VE-cadherin. DAPI overlay is shown for VE-cadherin in the control culture. (C) Representative flow cytometry histograms of VE-cadherin expression in control and RA-treated cultures. Mean ± S.D. of VE-cadherin+ cells due to RA treatment was calculated from seven biological replicates where a biological replicate is defined as an independent hPSC differentiation experiment. (D) Western blots after treatment with 0, 1, or 10 μM RA. (E) Representative flow cytometry dot plots of GLUT-1 and PECAM-1 populations in control and RA-treated differentiating cultures. Red dots, PECAM-1+/GLUT-1+ cells; blue dots, PECAM-1+/GLUT-1− cells; green dots, PECAM-1−/GLUT-1− cells. Statistical significance listed in the Results section was calculated on three independent biological replicates using the student's unpaired t-test. (F) Total cell number was quantified by counting trypsin-dissociated cells. Control cells, N = 18; RA-treated cells, N = 7. Data are represented as mean ± S.D. with statistical significance calculated using the student's unpaired t-test.
Next, the effect of RA treatment on differentiation efficiency was tested by evaluating the percentage of the mixed differentiating cultures that acquired BMEC properties as defined by PECAM-1+ cells having elevated GLUT-1 expression (GLUT-1+)18. The percentage of PECAM-1+ cells did not change upon RA treatment (control: 66 ± 6% versus RA-treated cells: 67 ± 15%, p > 0.05) (Fig. 1E). RA treatment also did not affect the fraction of the endothelial cells becoming PECAM+/GLUT-1+ BMECs (control: 88 ± 6% versus RA-treated: 82 ± 16%, p > 0.05) (Fig. 1E). While RA treatment did not change the overall distribution of BMECs in the culture dish, it did result in a statistically significant increase (195 ± 70%; p < 0.0001) in total cell number (Fig. 1F). Therefore, by increasing the proliferation of both BMECs and other cells in the dish (e.g. neural progenitors) without changing their relative abundance, RA treatment led to an approximately 2-fold increase in BMEC yield.
Given the initial findings that adherens and tight junction protein expression were enhanced by RA addition during BMEC differentiation, we sought to determine if RA effects continued to manifest themselves in purified BMEC cultures18. Several important measures of a robust BBB phenotype, including smooth tight junction strands7,29, elevated TEER and active efflux transport systems were therefore evaluated. Control and RA-treated iPSC-derived BMECs were purified by selective adhesion on collagen/fibronectin-coated polystyrene plates or Transwell filters and grown to confluence as monocultures in EC medium with or without RA, as appropriate (Fig. 2A). As previously demonstrated for control BMECs18, RA-treated BMECs could be purified by passage onto collagen/fibronectin and flow cytometry analysis demonstrated that 95–98% of the subcultured cells expressed VE-cadherin, claudin-5 and occludin (Fig. 2B). Immunocytochemical analysis also indicated that claudin-5 and occludin localized to cell-cell borders in both control and RA-treated BMECs (Fig. 2C). Quantitative morphological analysis of tight junction continuity indicated that control iPSC-derived BMECs possessed a percentage of discontinuous tight junction strands with frayed edges (13 ± 7%), while RA-treated BMECs displayed almost entirely smooth junctions (2 ± 1% frayed edges; Fig. 2C, p < 0.05). Once confluent, purified iPSC-derived BMECs were shifted to modified EC medium (EC medium lacking bFGF) and TEER measurements were used to functionally evaluate the tight junctions. Similar to our previous results, control iPSC-derived BMECs demonstrated TEER values of 228 ± 57 Ωxcm2 (ref. 18). However, RA-treated BMECs exhibited strikingly increased TEER of 2,940 ± 800 Ωxcm2 (Fig. 2D) across 12 independent biological replicates (Table 1). Collectively, these data indicate that RA treatment during differentiation significantly enhances the barrier phenotype of purified iPSC-derived BMECs.

Passive and active barrier properties in purified RA-treated IMR90-4-derived BMECs.
10 μM RA was used for all experiments. (A) Analysis was conducted on purified BMECs in 12-well plates or Transwell filters with or without RA treatment. (B) Flow cytometric purity analysis of VE-cadherin, claudin-5 and occludin expressing cells in purified RA-treated BMEC preparations. (C) Evaluation of tight junction continuity for control and RA-treated BMECs. Frayed tight junction strands (arrowheads) were quantified. (D) TEER values for control and RA-treated BMECs. Statistical significance was calculated using the student's unpaired t-test. Mean ± S.D. was calculated across biological replicates (see Table 1 for full details). (E) Changes in efflux transporter gene expression by qPCR. ABCB1, ABCG2, ABCC1, ABCC2, ABCC5 and STRA6 were upregulated due to RA treatment (positive change in ΔΔCt). ABCC4 and SLC2A1 were unaffected by RA. Mean ± S.D. were calculated from triplicate reactions and results are representative of two biological replicates. Statistical significance was calculated using the student's unpaired t-test (*, p < 0.01; **, p < 0.005). (F) Immunocytochemical detection of p-glycoprotein, BCRP and MRP1 in RA-treated BMECs. DAPI nuclear stain is overlaid in blue. (G) Efflux transporter inhibition in control or RA-treated BMECs. All inhibitor-treated samples (control or RA-treated) were independently normalized to the corresponding non-inhibitor-treated samples. Statistical significance was calculated using the student's unpaired t-test and is discussed in the Results section. Only normalized DCFDA uptake in the presence of MK 571 was significantly different between control and RA-treated BMECs (p < 0.005).
RA was next examined for its capability to modulate transporter expression and activity. Transcripts encoding ABCB1 (p-glycoprotein), ABCG2 (breast cancer resistance protein, BCRP), ABCC1 (multidrug resistance protein 1, MRP1), ABCC2 (MRP2), ABCC5 (MRP5) and STRA6 were upregulated following RA treatment (ΔΔCt values of 1.6 ± 0.4, 1.8 ± 0.2, 2 ± 0.7, 1.3 ± 0.3, 0.9 ± 0.3 and 2.5 ± 0.7 respectively, compared to control) while ABCC4 (MRP4) and SLC2A1 (GLUT-1) did not exhibit any change (Fig. 2E). P-glycoprotein, BCRP and MRP1 were detected by immunocytochemistry in RA-treated iPSC-derived BMECs (Fig. 2F) and flow cytometric quantification demonstrated that RA treatment increased MRP1 protein expression 1.3-fold while p-glycoprotein and BCRP protein expression levels were unchanged (Supplementary Fig. S2 online). To assess changes in efflux transporter activity resulting from RA treatment, purified iPSC-derived BMECs were first incubated with the cell-permeable, fluorescent p-glycoprotein substrate rhodamine 123 and intracellular accumulation was measured in the presence or absence of cyclosporin A (CsA), a p-glycoprotein inhibitor. Both control and RA-treated BMECs exhibited an increase in rhodamine 123 accumulation in the presence of CsA (control, 221 ± 39%; RA, 316 ± 117%; Fig. 2G) but the normalized increases between each treatment were not statistically different (p > 0.05). Similarly, if rhodamine 123 transport was measured across an iPSC-derived BMEC monolayer cultured on a Transwell filter, increased apical to basolateral transport was observed in the presence of CsA (control, 172 ± 7%; RA, 187 ± 30%; Fig. 2G) but the relative increase in the RA treated system was again statistically insignificant (p > 0.05). These results indicate p-glycoprotein activity was not increased by RA treatment. Next, the effect of RA treatment on the accumulation of radiolabeled doxorubicin, a pan-substrate of p-glycoprotein, BCRP and the MRP family, was tested. Treatment with CsA, Ko143 (a BCRP inhibitor), or MK 571 (an MRP family inhibitor) resulted in increased accumulation (Fig. 2G) but once again, the relative increase between control and RA-treated BMECs was statistically insignificant (p > 0.05 for all inhibitors). Thus, although flow cytometric quantification indicated increased MRP1 protein levels upon RA treatment, doxorubicin accumulation in the presence of MRP inhibitor was not significantly enhanced by RA. However, with a pan-substrate such as doxorubicin, competing efflux by p-glycoprotein and BCRP efflux could mask any putative increase in MRP activity. As such, we tested accumulation of carboxymethyl-2′,7′-dichlorofluorescein diacetate (DCFDA), a cell-permeable MRP family substrate that becomes fluorescent upon acetate cleavage by intracellular esterases. As expected, control BMECs demonstrated an increase in uptake upon MK 571 treatment (164 ± 7%) but contrasting to the results observed with doxorubicin, RA-treated BMECs demonstrated significantly higher levels of uptake in the presence of MK 571 (436 ± 57%; p < 0.005; Fig. 2G). Collectively, these results indicate that of the three efflux transporter families tested, only MRP expression and activity were increased by RA treatment during the iPSC differentiation process.
RA effects translate to BMEC differentiation from other hPSC lines
Having demonstrated improved BBB properties in response to RA using IMR90-4 iPSCs, we sought to confirm RA-specific effects on other hPSC lines. Similar to the IMR90-4 line, differentiating DF-19-9-11T iPSC cultures exhibited induced VE-cadherin expression in response to RA treatment (Supplementary Fig. S3A online). RA-treated DF19-9-11T-derived BMECs possessed smooth and continuous tight junctions after purification (Supplementary Fig. S3B and S3C online) and exhibited an elevated TEER of 1,710 ± 420 Ωxcm2 (Table 1 and Supplementary Fig. S3D online).
We next tested H9 hESCs, which in our previous work could be differentiated to cells expressing BMEC markers but did not generate pure barrier-forming endothelium18. This contrasts with the IMR90-4 iPSC line that yielded a high fraction of BMECs during differentiation, even in the absence of RA (Fig. 1E)18. We therefore tested whether RA could enhance BMEC formation at the expense of non-BBB endothelium during H9 hESC differentiation. Addition of RA during 2 days of EC medium treatment or an extended 3 days of EC medium treatment was found to induce VE-cadherin expression (Supplementary Fig. S4A online) but did not the change the fraction of PECAM-1+ cells becoming BMECs (Supplementary Fig. S4B online). If instead, 1 day of EC medium expansion was followed by 3 days of culture in EC medium containing RA (6D UM, 1D EC, 3D EC+RA), the number of PECAM-1+/GLUT-1+ BMECs increased to nearly 95% of the total PECAM-1+ population (Supplementary Fig. S4B online). These BMECs could be purified by passaging onto collagen/fibronectin (Supplementary Fig. S4C online), which led to elevated TEER (646 ± 310 Ωxcm2) compared to BMECs receiving 4D EC treatment without RA (6D UM, 4D EC; 123 ± 4 Ωxcm2; p < 0.05; Supplementary Fig. S4D online). The increased TEER was also accompanied by smooth tight junctions in the RA-treated BMECs (Supplementary Fig. S4E online). Thus, extended EC treatment plus RA supplementation (1D EC, 3D EC+RA) can be used to induce BBB phenotypes in lines that may display deficits in BMEC differentiation efficiency using the standard 2D EC medium culture protocol.
iPSC-derived BMECs respond to inductive cues from NPC-derived astrocytes and neurons and primary human brain pericytes
We previously demonstrated that human NPCs differentiated for 12 days could elevate TEER values in primary rat BMECs upon co-culture7. To probe whether iPSC-derived BMECs would also respond to co-cultured human NPC-derived astrocytes and neurons, primary human NPCs were differentiated for 9, 12, or 15 days to form mixtures of neurons and astrocytes (Supplementary Fig. S5A online) and co-culture with IMR90-4-derived BMECs (Fig. 3A, -RA, Scheme MNPC) was initiated in medium containing 10% FBS (denoted NPC medium). All co-cultures produced elevated TEER (>400 Ωxcm2) and no statistically significant differences were observed with respect to NPC differentiation time (Fig. 3B). By comparison, monoculture controls (Fig. 3A, -RA, Scheme MM) only reached a peak TEER of 108 ± 3 Ωxcm2. IMR90-4-derived BMEC co-culture with primary human brain pericytes (Supplementary Fig. S5 online and Fig. 3A, -RA, Scheme MP) was then compared to co-culture with 12-day differentiated NPCs (Fig. 3A, -RA, Scheme MNPC) and while pericytes increased TEER, co-culture with differentiated NPCs induced higher TEER values (Fig. 3C; 297 ± 36 Ωxcm2 versus 435 ± 25 Ωxcm2; p < 0.01). We next tested whether pericytes could “prime” the hPSC-derived BMECs for NPC co-culture by culturing subconfluent hPSC-derived BMECs with pericytes immediately following purification and then transferring the confluent BMECs into co-culture with differentiated NPCs 24 hours later (Fig. 3A, -RA, Scheme PNPC). This sequential condition yielded the highest TEER compared with either pericyte or 12-day differentiated NPC co-culture alone (Fig. 3C; 611 ± 63 Ωxcm2; p < 0.02). Thus, maximum TEER induction occurred when BMECs were exposed to both pericytes and differentiated NPCs.

Effects of pericyte and differentiated NPC co-culture on TEER induction in IMR90-4-derived BMECs.
(A) Description of co-culture schemes. For all experiments, IMR90-4-derived BMECs were purified into standard EC medium for the subculture phase and switched to NPC medium for the co-culture phase. (B) NPCs were differentiated for 9, 12, or 15 days prior to co-culture with IMR90-4-derived BMECs (Scheme MNPC). Monocultured IMR90-4-derived BMECs were used as a negative control (Scheme MM). (C) TEER measured on IMR90-4-derived BMECs in co-culture with pericytes and differentiated NPCs (Schemes MP, MNPC and PNPC). *, p < 0.01 between Schemes MP and MNPC; **, p < 0.02 between Schemes MNPC and PNPC. (D) 10 μM RA treatment prior to pericyte and differentiated NPC co-culture. TEER for RA-treated BMECs was measured upon confluence (0 h) and after 24 hours (24 h). The increase in baseline TEER at confluence for Schemes PP and PNPC compared to Schemes MM, MP and MNPC was reproducible across six biological experiments. For all experiments, triplicate filters were used to calculate mean ± S.D. and statistical significance was calculated using the student's unpaired t-test.
Co-culture enhances barrier properties of RA-treated hPSC-derived BMECs
To determine if RA treatment could synergize with the inductive tightening effects of pericytes and NPC-derived astrocytes and neurons, RA-treated hPSC-derived BMECs were incorporated into the human co-culture models illustrated in Figure 3A using NPC medium during the co-culture phase. As was observed with control IMR90-4-derived BMECs, co-culture of RA-treated BMECs with pericytes (Fig. 3A, +RA, Scheme MP) increased TEER above the monoculture control (Fig. 3D and Table 2; t = 24 h, 1,600 ± 53 Ωxcm2 vs. 1,040 ± 110 Ωxcm2; p < 0.002) while co-culture with differentiated NPCs (Fig. 3A, +RA, Scheme MNPC) increased TEER more substantially (Fig. 3D; t = 24 h, 2,370 ± 120 Ωxcm2; p < 0.001). If RA-treated BMECs were primed with pericytes during the subculture phase (Fig. 3A, +RA, Schemes PP and PNPC), TEER at confluence was significantly enhanced compared to monocultured BMECs (Fig. 3D; t = 0 h, 2,070 ± 120 Ωxcm2 vs. 580 ± 42 Ωxcm2; p < 0.0001). Co-culture of these BMECs with differentiated NPCs further enhanced TEER compared to BMECs that remained in co-culture with pericytes (Fig. 3D; +RA, Scheme PNPC vs. PP; t = 24 h, 3,370 ± 150 Ωxcm2 vs. 2,700 ± 53 Ωxcm2; p < 0.002). To evaluate the reproducibility of the optimal PNPC co-culture for barrier maturation, 7 independent cultures of IMR90-4-derived BMECs were subcultured with pericytes and subsequently co-cultured with 7 independent sets of NPC-derived progeny pre-differentiated for a range of time (10–19 days) and TEER exceeded at least 2,500 Ωxcm2 in each experiment (Supplementary Fig. S6 online). Thus, the model was quite reproducible with regards to independent iPSC and NPC differentiation experiments.
Optimization of co-culture using modified EC medium
The use of 10% FBS-containing NPC medium during the co-culture phase was based on our previous studies7,18. However, when employing EC medium that lacked exogenous bFGF (modified EC medium), monocultured RA-treated BMECs could achieve a similarly elevated TEER to that of the PNPC co-culture system (Fig. 2D compared to Fig. 3D; ~3,000 Ωxcm2). Therefore, we replaced NPC medium in the various co-culture set-ups with modified EC medium and re-evaluated TEER induction (Fig. 4A). Pericyte co-culture in modified EC medium (Fig. 4A, Scheme PP) yielded TEER up to 4,450 ± 170 Ωxcm2 whereas a switch to differentiated NPC co-culture (Fig. 4A, Scheme PNPC) enhanced TEER further to a maximum at 24 hours (Fig. 4B; 5,160 ± 320 Ωxcm2; p < 0.05). If a non-neural control (primary human foreskin fibroblasts; Fig. 4A, Scheme FF) was used in place of pericytes for both the subculture and co-culture phases, TEER was also somewhat elevated above monoculture (Scheme MM) upon reaching confluence (Fig. 4B at 0 h; 2,110 ± 30 Ωxcm2; p < 0.01) but not after 24 hours (Fig. 4B; 2,470 ± 120 Ωxcm2 vs. 2,410 ± 370 Ωxcm2; p > 0.05) and remained well below the TEER induced by pericytes. The optimum PNPC co-culture scheme in modified EC medium reached TEER of >5,000 Ωxcm2 for 5 out of 6 independent differentiation and co-culture experiments with a maximum value of 5,350 ± 250 Ωxcm2, again proving to be independent of the NPC differentiation timeframe (Fig. 4C and Table 2). Moreover, the TEER remained elevated above 3,000 Ωxcm2 for over 3 days (Fig. 4B). Importantly, under these same optimal co-culture and medium conditions, DF19-9-11T-derived BMECs reached TEER values of 4,740 ± 300 Ωxcm2 and H9-derived BMECs attained TEER values of 1,680 ± 95 Ωxcm2 (Table 2), indicating that the PNPC co-culture scheme is generalizable across different hPSC lines. As expected, the extremely high TEER reached by IMR90-4-derived BMECs correlated with high fidelity tight junctions (Fig. 4D; 1.4 ± 0.6% frayed junctions), although tight junction fidelity was not significantly different from a direct comparison to BMECs only treated with RA (Fig. 4D; 1.5 ± 0.9% frayed junctions; p > 0.05). Finally, to measure the general barrier restrictiveness to paracellular flux, the permeability (Pe) of radiolabeled sucrose was found to be very low and indistinguishable between RA-treated BMECs alone (5.3 ± 1.7 × 10−7 cm/s) and the optimal PNPC system (5.7 ± 1.2 × 10−7 cm/s), which despite the TEER difference (~3,000 vs. ~5,000 Ωxcm2) agrees with previous reports demonstrating negligible differences in passive permeability of small molecules above certain TEER thresholds7,30. Taken together, the combination of RA treatment, pericyte and differentiated NPC co-culture and media optimization leads to a human BBB model with very tight barrier properties.
Discussion
The purpose of this work was to construct a renewable, robust human BBB multicellular co-culture model employing hPSCs, NPCs and pericytes. Using previous studies as guides25,26,31, RA was identified as a significant modulator of BMEC properties during hPSC differentiation that greatly enhanced physical barrier characteristics as demonstrated by elevated TEER in BMECs cultured alone or with neurovascular cell co-culture. In recent work, RA treatment on the hCMEC/D3 human brain endothelial cell line served to increase occludin and VE-cadherin expression and the authors suggested that RA secreted by radial glia may be involved in BBB development27. In our study, when RA was added during the endothelial progenitor expansion phase of hPSC-derived BMEC differentiation, similar results were observed including an earlier onset of VE-cadherin expression and increased occludin expression. Moreover, the BMEC yield was increased 2-fold and the tightness of the hPSC-derived BMEC monolayers as measured by elevated TEER was significantly enhanced for three different hPSC lines. Somewhat unexpectedly, RA treatment resulted in decreased claudin-5 expression. However, the Western blotting analysis was conducted using whole-cell lysates and does not take into account the substantially improved intercellular claudin-5 junctional continuity upon RA treatment (Fig. 2C). We and others have previously observed a strong correlation between such junctional continuity and resultant barrier phenotype6,29,32. In addition, previous work has demonstrated claudin-5 expression is relatively constant across peripheral and BBB endothelium while occludin expression is increased at the BBB relative to other vascular beds31. Thus, a combination of claudin-5 localization and elevated occludin expression may be the key phenotypic indicators of increased barrier function31,33. RA treatment of hPSC-derived BMECs also selectively increased MRP efflux activity, which agrees with reports demonstrating that signaling via nuclear receptors can regulate efflux transporter expression and activity at the blood-brain barrier in vitro and in vivo34,35,36,37. RA influences many aspects of brain development, such as anterior/posterior axis patterning in the hindbrain and anterior spinal cord38,39,40 and regulation of neurogenesis41,42,43. During BMEC differentiation, RA could trigger several modes of action. RA may act directly on the developing endothelial cells to upregulate BBB properties, it could induce changes in the neural cells to indirectly promote BBB differentiation, or it could act by a combination of these mechanisms. Future work will be necessary to deconvolute the RA signaling mechanisms affecting the hPSC-derived BMEC differentiation scheme.
While RA treatment significantly affected resultant BMEC phenotype, RA treatment did not have a large impact on the percentage of endothelial cells becoming BMECs for the IMR90-4 iPSC line, as this line already yields relatively pure BMEC populations in the absence of RA. In contrast, for the H9 hESC line, which forms many endothelial cells devoid of BBB characteristics in the absence of RA, supplementation with RA in conjunction with optimized differentiation timing was capable of enhancing BMEC formation such that it could form a substantial barrier upon purification. Taken together, RA provided a substantial, generalizable inductive effect during the hPSC-derived BMEC differentiation process that improved the reliability and quality of the resultant BMECs. While our results indicate that some hPSC line-to-line differentiation optimization is required, these RA-based methods could in principle be used to better facilitate the generation of BBB models using hESC and iPSC lines derived in other laboratories. The capability to translate this work to iPSC lines derived from diseased patients could in turn open new avenues for iPSC-based in vitro modeling of complex neurological diseases that may feature a BBB component44.
After analyzing the effects of RA during hPSC differentiation to BMECs, co-culture of RA-treated BMECs with differentiated NPCs was tested for further improvement in barrier function. We had previously shown that the duration of rat NPC differentiation (12 days) was critical in achieving neurons and astrocytes capable of inducing BBB properties in primary rat BMECs7. Furthermore, human NPCs differentiated for 12 days induced similar BBB properties as rat NPCs7, which led us to test human NPC differentiation times both above and below 12 days to determine if an optimum existed. NPCs differentiated for 9–24 days exhibited no significant differences in their ability to upregulate TEER in IMR90-4 iPSC-derived BMECs, suggesting a minimal NPC differentiation time is sufficient to generate neuron/astrocyte mixtures capable of modulating TEER. Importantly, the general insensitivity of BBB properties in the hPSC-derived BMECs relative to the maturation of NPC-derived progeny greatly simplifies the alignment of hPSC and NPC differentiation timelines thereby facilitating the use of hPSC/NPC co-culture models.
We also explored the inclusion of pericytes in this BBB model prior to co-culture with NPC-derived progeny because pericytes share the capillary basement membrane with endothelial cells while playing important roles in endothelial maturation and survival45 and pericytes contribute to BBB properties in the developing brain prior to the onset of astrocyte differentiation9. Expansion of purified hPSC-derived BMECs in the presence of pericytes, followed by co-culture with differentiated NPCs, more effectively enhanced barrier function (TEER) than co-culture with either cell type alone. These findings are similar to those of rodent in vitro BBB studies that employed various co-culture combinations of primary isolated pericytes, astrocytes and BMECs4,5. Overall, this sequential tri-culture system constitutes a human BBB model constructed entirely from highly scalable sources. Pericytes have previously been cultured for twenty weeks with over forty population doublings46, which demonstrates their proliferative capacity, although ideally in the future, these may also be derived from stem cells47. Similarly, NPCs have long been recognized for their extensive self-renewal capabilities48, leading to a virtually unlimited supply of neurons and astrocytes upon differentiation. Thus, unlike models which require continuous isolations of primary cells, this model derives all of its cellular components from sources that can be propagated in vitro, which helps simplify model implementation.
Perhaps the most striking result from this study is the absolute TEER that can be achieved in this model, as a key to using BBB models for molecular transport assays is the overall tightness of the paracellular junctions in the BMEC monolayer. Treatment with RA alone produced TEER above 3,000 Ωxcm2 in monocultured hPSC-derived BMECs and the combination of RA treatment with sequential pericyte and differentiated NPC co-culture resulted in hPSC-derived BMECs possessing a maximum TEER in excess of 5,000 Ωxcm2, which is several-fold higher than the tightest animal-derived BBB models and more than 10-fold higher than any published human model3. While these dramatic increases in tightness were initially unexpected, there is a precedent for this level of BBB tightness in vivo. Classic experiments performed by Crone and Olesen measured an average TEER of 1,870 Ωxcm2 at the frog BBB with a maximum TEER value of 2,976 Ωxcm2 and using theoretically calculated values of conductance, they suspected the true maximum TEER value to be around 4,000 Ωxcm2 (ref. 49). Experiments on the brains of maturing rats (above 21 days of gestation) by Butt and co-workers measured an average TEER of only 1,490 ± 170 Ωxcm2 in brain arterial vessels and 918 ± 127 Ωxcm2 in venous vessels, but they also measured a maximum value of 5,900 Ωxcm2 and stated in their report that “any potential deterioration of the preparation would tend to lower the measured values, so it is conceivable that the higher figures reflect the true resistance of the blood-brain barrier”22. It should also be noted that these measurements were obtained for vessels at the pial surface and parenchymal vessels may possess higher resistance values. A study by Smith and Rapoport supports this notion by estimating an in vivo TEER of 8,000 Ωxcm2 at the rat BBB based on measured permeability coefficients of radioisotopic ions50. Overall, it is likely that the TEER values achieved in the RA-treated hPSC-derived BBB model are approaching relevant TEER values found in vivo. The longstanding issue of relatively leaky monolayers has diminished the overall utility of human BBB models13 and the extreme tightness of hPSC-derived BMEC models using the methods described herein promises to be advantageous for many BBB applications.
Methods
hPSC differentiation to BMECs
IMR90-4 and DF19-9-11T iPSCs and H9 hESCs were maintained between passages 26–42 on Matrigel (BD Biosciences) in mTeSR1 medium (STEMCELL Technologies) or on irradiated mouse embryonic fibroblasts (MEFs) in standard unconditioned medium (UM) as previously described18. For differentiation, cells were passaged onto Matrigel in mTeSR1 medium for 2–3 days of expansion and then switched to unconditioned medium (UM) lacking bFGF for 6 days. Human endothelial serum-free medium (hESFM; Life Technologies) supplemented with 20 ng/mL bFGF (R&D Systems) and 1% platelet-poor plasma derived bovine serum (Biomedical Technologies, Inc.) was then added for an additional 2–4 days. All-trans RA (Sigma) was reconstituted in DMSO and included at concentrations of 1–10 μM depending on the experiment. Cells were then dissociated with Versene (Life Technologies) and plated onto 12-well tissue culture polystyrene plates or 1.12 cm2 Transwell-Clear® permeable inserts (0.4 μm pore size) coated with a mixture of collagen IV (400 μg/mL; Sigma) and fibronectin (100 μg/mL; Sigma) in H2O. Culture plates were incubated with the coating for at least 30 min at 37°C, while the inserts were incubated for a minimum of 4 h at 37°C. Resultant, purified hPSC-derived BMECs were then grown in EC medium for 24 h (with or without RA); in some experiments, primary pericytes or fibroblasts were co-cultured with BMECs during these 24 h (see description below). After this 24 h period, BMECs were continued as monoculture or co-cultured as described below. In our previous publication, we had utilized dispase for subculturing the BMECs18, but non-enzymatic treatment of the BMECs with Versene results in less debris attached to the BMEC monolayer. We had also used hPSCs exclusively maintained on MEFs prior to differentiation18, but in this study no noticeable differences in BBB properties were observed between hPSCs maintained on MEFs and hPSCs maintained under feeder-independent conditions and we now exclusively use hPSCs maintained in mTeSR1 on Matrigel.
Primary cell culture: human neural progenitor cells, human pericytes and human foreskin fibroblasts
Human neural progenitor cells (NPCs) were maintained as previously described7 in NPC culture medium (70%:30% DMEM/F12 (Sigma/Life Technologies) supplemented with 2% B27 (Life Technologies), 1% antibiotic-antimycotic (Life Technologies), 20 ng/mL bFGF, 20 ng/mL epidermal growth factor (EGF; Sigma), 10 ng/mL leukemia inhibitory factor (LIF; Millipore) and 5 μg/mL heparin (Sigma)) and cells were passaged every 7–10 days using standard chopping methods. To initiate differentiation, NPCs were dissociated with accutase (Life Technologies) and seeded onto 12-well plates or filters coated with poly-L-lysine/laminin (Sigma) at a density of 2 × 105 cells/well or 5 × 104 cells/filter. Differentiation medium consisted of NPC culture medium with the growth factors replaced by 1% fetal bovine serum (FBS; Life Technologies). Medium was changed every third day. NPCs were differentiated 9–24 days prior to use in co-culture experiments.
Primary fetal human brain pericytes (Sciencell) were maintained in DMEM supplemented with 10% FBS and expanded for two passages, upon which stock vials were frozen in liquid nitrogen. Pericytes were then utilized from the original cell culture, or from thawed stock vials, between passages 2–12. Medium was changed every second day and cells were subcultured after reaching ~90% confluency. For subculture, pericytes were washed once with PBS and incubated with accutase for 5–10 min until cells began to detach, then re-seeded at a density of 5 × 103 cells/cm2 on poly-L-lysine-coated flasks, plates, or filters. Pericytes were seeded in 12-well plates or filters 1–2 days prior to co-culture and typically were 50–80% confluent when co-culture was initiated. Primary human foreskin fibroblasts (BJ line; ATCC) were cultured in Minimum Essential Medium (Sigma) supplemented in 10% FBS and used as a negative control in certain co-culture experiments.
Initiation of co-culture experiments
The timing of co-culture is shown in each relevant figure. During the 24 hour subculture phase, hPSC-derived BMECs were monocultured or co-cultured with pericytes or fibroblasts in EC medium (with or without RA). After the 24 h subculture phase, hPSC-derived BMECs were maintained as a monoculture or co-cultured with differentiated NPCs, pericytes, or fibroblasts. Depending on the experiment, co-culture was conducted in NPC differentiation medium containing 10% FBS (denoted NPC medium in the Results section) or hESFM containing 1% PDS but not bFGF (denoted modified EC medium). Trans-endothelial electrical resistance (TEER) was measured immediately prior to co-culture using an EVOM voltohmmeter with STX2 electrodes (World Precision Instruments) and approximately every 24 h thereafter as necessary. Raw TEER values were adjusted by subtracting TEER measured across an empty filter and multiplied by filter surface area to yield TEER (Ωxcm2) imparted by the BMECs. Each filter was measured three times and triplicate filters were used for each culture condition. Because TEER can fluctuate with temperature, care was taken to assure measurements were taken at 37°C.
Immunocytochemistry and analysis of tight junction fidelity
Immunocytochemistry was conducted as previously described with minor modifications18. Cells were blocked in PBS containing 40% goat serum or donkey serum (40% PBSG or 40% PBSD; Sigma) at room temperature for 30 min, depending on the species of primary antibody. 0.1% Triton X-100 (TX-100) was included if membrane permeabilization was required. Primary antibodies are listed in Supplementary Table S1. Images were taken with an Olympus epifluorescence microscope using a Diagnostic Instruments camera run by MetaVue software. For quantitative analysis of BMEC integrity, the percentage of cells expressing frayed tight junctions was counted using BMECs immunolabeled for occludin. Cells were defined as having frayed tight junctions if any cell-cell contact point appeared discontinuous. A minimum of four separate frames and 1000 total cells were counted to obtain a percentage of frayed tight junctions.
Quantitative polymerase chain reaction (qPCR)
Cells were washed once with PBS and dissociated with accutase. Total RNA was extracted using an RNeasy Mini Kit (Qiagen) according to the manufacturer's instructions and quantified using a NanoDrop® ND-1000. cDNA was generated from 1 μg of total RNA using Omniscript reverse transcriptase (Qiagen) and an oligo-dT primer (Life Technologies). qPCR was conducted using 1 μL of cDNA and iQ SYBR Green Mastermix (Bio-Rad) on an iCycler (Bio-Rad). Relative expression was quantified between samples using the comparative cycle threshold (Ct) method with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as the housekeeping gene. Primer sequences were exactly as previously reported18.
Flow cytometry
Cells were washed once with PBS and dissociated with accutase for 5 min, then spun down and fixed in 2% paraformaldehyde for 15 min or 100% methanol for 10 min. Cells were blocked and permeabilized in 40% PBSG or 10% PBSD with or without 0.1% TX-100 for 20 min at room temperature and then incubated with primary antibody diluted in 40% PBSG or 10% PBSD for 1 h at room temperature or overnight at 4°C. Antibodies are listed in Supplementary Table S1 and mouse, rabbit, or goat IgG isotype controls were employed at matching concentration. After being washed twice with PBS containing 5% FBS, cells were incubated with goat anti-rabbit Alexa Fluor 488, goat anti-mouse Alexa Fluor 647, donkey anti-goat Alexa Fluor 488, or donkey anti-mouse Alexa Fluor 488 (1:200 dilution) for 30 min at room temperature. After another two washes with PBS containing 5% FBS, cells were analyzed on a FACScaliber flow cytometer and the IgG controls were used to quantify positive labeling. GLUT-1+ cells were quantified using a GLUT-1/forward scatter plot referenced to basal GLUT-1 expression measured at day 4 of UM culture as previously described18.
Western blots
Cells were washed once with PBS and lysed with RIPA buffer (Pierce). Protein concentration was quantified via BCA assay (Pierce) and proteins were then resolved by SDS-PAGE on 4–20% Tris-Glycine gradient gels (Life Technologies). After transfer to nitrocellulose membranes, blocking was conducted for 1 h in Tris-buffered saline (10 mM Tris-HCl, 100 mM NaCl, pH 7.5) containing 0.1% Tween-20 (TBST) and 5% milk. Samples were probed overnight at 4°C with anti-VE-cadherin (1:200), anti-claudin-5 (1:250), anti-occludin (1:1,000) and anti-beta-actin (Santa Cruz Biotechnology; 1:5,000) antibodies diluted in TBST with 5% milk. After being washed five times with TBST, samples were incubated with a peroxidase-conjugated anti-mouse secondary antibody (Life Technologies; 1:2,500) for 1 h at room temperature. Protein levels were detected via a SuperSignal West Pico Chemiluminescent Substrate (Pierce).
Efflux transporter and permeability assays
For accumulation assays, IMR90-4-derived BMECs differentiated in the presence or absence of RA were passaged onto 12-well plates in EC medium with 10 μM RA or an equivalent volume of DMSO. After reaching confluence, cells were incubated with 10 μM rhodamine 123 (Sigma), 0.25 μCi [14C]doxorubicin (PerkinElmer), or 10 μM carboxymethyl-2′,7′-dichlorofluorescein diacetate (DCFDA; Life Technologies) in transport buffer (distilled water with 0.12 M NaCl, 25 mM NaHCO3, 3 mM KCl, 2 mM MgSO4, 2 mM CaCl2, 0.4 mM K2HPO4, 1 mM HEPES and 0.1% bovine serum albumin (BSA; Sigma)) for 1 h at 37°C on a rotating platform. For inhibition experiments, cells were pre-incubated with 10 μM cyclosporin A (CsA; Sigma), 1 μM Ko143 (Sigma), or 10 μM MK571 (Sigma), depending on the experiment, for 1 h at 37°C on a rotating platform, then co-incubated with the inhibitor and compound of interest. At the conclusion of the experiment, cells were washed three times with PBS and lysed with PBS containing 5% TX-100. Fluorescence (485 nm excitation and 530 nm emission) was measured using a plate reader and radioactivity was measured on a scintillation counter. Fluorescence/radioactivity was normalized on a per cell basis by counting dissociated cells from parallel unlysed wells on a hemacytometer. For rhodamine 123 transport assays, IMR90-4-derived BMECs were seeded onto filters in EC medium with RA or DMSO and allowed to reach confluence over 24 h. After CsA pre-incubation, rhodamine 123 with or without CsA was added to the upper chamber in transport buffer and cells were incubated for 1 h at 37°C on a rotating platform. Aliquots were then extracted from the bottom chamber and fluorescence was quantified on a plate reader. To measure sucrose permeability, 0.4 μCi of [14C]sucrose was added to the upper chamber in transport buffer and cells were incubated for 1 h at 37°C on a rotating platform with aliquots drawn from the bottom chamber every 15 min and replaced with fresh buffer. The rate of accumulation of radioactive ligand was used to calculate Pe values. For assays with inhibitors, monolayer fidelity was confirmed at the end of each experiment by measuring TEER.
References
Zlokovic, B. V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 57, 178–201 (2008).
Pardridge, W. M. The blood-brain barrier: bottleneck in brain drug development. NeuroRx 2, 3–14 (2005).
Deli, M. A., Abraham, C. S., Kataoka, Y. & Niwa, M. Permeability studies on in vitro blood-brain barrier models: physiology, pathology and pharmacology. Cell Mol Neurobiol 25, 59–127 (2005).
Nakagawa, S. et al. A new blood-brain barrier model using primary rat brain endothelial cells, pericytes and astrocytes. Neurochem Int 54, 253–263 (2009).
Nakagawa, S. et al. Pericytes from brain microvessels strengthen the barrier integrity in primary cultures of rat brain endothelial cells. Cell Mol Neurobiol 27, 687–694 (2007).
Weidenfeller, C., Svendsen, C. N. & Shusta, E. V. Differentiating embryonic neural progenitor cells induce blood-brain barrier properties. J Neurochem 101, 555–565 (2007).
Lippmann, E. S., Weidenfeller, C., Svendsen, C. N. & Shusta, E. V. Blood-brain barrier modeling with co-cultured neural progenitor cell-derived astrocytes and neurons. J Neurochem 119, 507–520 (2011).
Daneman, R. et al. Wnt/beta-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc Natl Acad Sci U S A 106, 641–646 (2009).
Daneman, R., Zhou, L., Kebede, A. A. & Barres, B. A. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 468, 562–566 (2010).
Kuhnert, F. et al. Essential regulation of CNS angiogenesis by the orphan G protein-coupled receptor GPR124. Science 330, 985–989 (2010).
Lee, S. W. et al. SSeCKS regulates angiogenesis and tight junction formation in blood-brain barrier. Nat Med 9, 900–906 (2003).
Wosik, K. et al. Angiotensin II controls occludin function and is required for blood brain barrier maintenance: relevance to multiple sclerosis. J Neurosci 27, 9032–9042 (2007).
Cecchelli, R. et al. Modelling of the blood-brain barrier in drug discovery and development. Nat Rev Drug Discov 6, 650–661 (2007).
Bernas, M. J. et al. Establishment of primary cultures of human brain microvascular endothelial cells to provide an in vitro cellular model of the blood-brain barrier. Nat Protoc 5, 1265–1272 (2010).
Rubin, L. L. et al. A cell culture model of the blood-brain barrier. J Cell Biol 115, 1725–1735 (1991).
Weksler, B. B. et al. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. Faseb J 19, 1872–1874 (2005).
Forster, C. et al. Differential effects of hydrocortisone and TNFalpha on tight junction proteins in an in vitro model of the human blood-brain barrier. J Physiol 586, 1937–1949 (2008).
Lippmann, E. S. et al. Derivation of blood-brain barrier endothelial cells from human pluripotent stem cells. Nat Biotechnol 30, 783–791 (2012).
Zenker, D., Begley, D., Bratzke, H., Rubsamen-Waigmann, H. & von Briesen, H. Human blood-derived macrophages enhance barrier function of cultured primary bovine and human brain capillary endothelial cells. J Physiol 551, 1023–1032 (2003).
Cohen-Kashi Malina, K., Cooper, I. & Teichberg, V. I. Closing the gap between the in-vivo and in-vitro blood-brain barrier tightness. Brain Res 1284, 12–21 (2009).
Patabendige, A., Skinner, R. A. & Abbott, N. J. Establishment of a simplified in vitro porcine blood-brain barrier model with high transendothelial electrical resistance. Brain Res 1521, 1–15 (2013).
Butt, A. M., Jones, H. C. & Abbott, N. J. Electrical resistance across the blood-brain barrier in anaesthetized rats: a developmental study. J Physiol 429, 47–62 (1990).
Kawaguchi, R. et al. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science 315, 820–825 (2007).
Bouillet, P. et al. Developmental expression pattern of Stra6, a retinoic acid-responsive gene encoding a new type of membrane protein. Mech Dev 63, 173–186 (1997).
El Hafny, B. et al. Modulation of P-glycoprotein activity by glial factors and retinoic acid in an immortalized rat brain microvessel endothelial cell line. Neurosci Lett 236, 107–111 (1997).
Lechardeur, D., Schwartz, B., Paulin, D. & Scherman, D. Induction of blood-brain barrier differentiation in a rat brain-derived endothelial cell line. Exp Cell Res 220, 161–170 (1995).
Mizee, M. R. et al. Retinoic acid induces blood-brain barrier development. J Neurosci 33, 1660–1671 (2013).
Xu, Y. et al. Revealing a core signaling regulatory mechanism for pluripotent stem cell survival and self-renewal by small molecules. Proc Natl Acad Sci U S A 107, 8129–8134 (2010).
Calabria, A. R., Weidenfeller, C., Jones, A. R., de Vries, H. E. & Shusta, E. V. Puromycin-purified rat brain microvascular endothelial cell cultures exhibit improved barrier properties in response to glucocorticoid induction. J Neurochem 97, 922–933 (2006).
Gaillard, P. J. & de Boer, A. G. Relationship between permeability status of the blood-brain barrier and in vitro permeability coefficient of a drug. Eur J Pharm Sci 12, 95–102 (2000).
Daneman, R. et al. The mouse blood-brain barrier transcriptome: a new resource for understanding the development and function of brain endothelial cells. PLoS One 5, e13741 (2010).
Weidenfeller, C., Schrot, S., Zozulya, A. & Galla, H. J. Murine brain capillary endothelial cells exhibit improved barrier properties under the influence of hydrocortisone. Brain Res 1053, 162–174 (2005).
Ohtsuki, S., Yamaguchi, H., Katsukura, Y., Asashima, T. & Terasaki, T. mRNA expression levels of tight junction protein genes in mouse brain capillary endothelial cells highly purified by magnetic cell sorting. J Neurochem 104, 147–154 (2008).
Durk, M. R. et al. 1alpha,25-Dihydroxyvitamin D3-liganded vitamin D receptor increases expression and transport activity of P-glycoprotein in isolated rat brain capillaries and human and rat brain microvessel endothelial cells. J Neurochem 123, 944–953 (2012).
Wang, X., Hawkins, B. T. & Miller, D. S. Aryl hydrocarbon receptor-mediated up-regulation of ATP-driven xenobiotic efflux transporters at the blood-brain barrier. Faseb J 25, 644–652 (2011).
Hartz, A. M., Mahringer, A., Miller, D. S. & Bauer, B. 17-beta-Estradiol: a powerful modulator of blood-brain barrier BCRP activity. J Cereb Blood Flow Metab 30, 1742–1755 (2010).
Wang, X., Sykes, D. B. & Miller, D. S. Constitutive androstane receptor-mediated up-regulation of ATP-driven xenobiotic efflux transporters at the blood-brain barrier. Mol Pharmacol 78, 376–383 (2010).
Maden, M., Gale, E., Kostetskii, I. & Zile, M. Vitamin A-deficient quail embryos have half a hindbrain and other neural defects. Curr Biol 6, 417–426 (1996).
Liu, J. P., Laufer, E. & Jessell, T. M. Assigning the positional identity of spinal motor neurons: rostrocaudal patterning of Hox-c expression by FGFs, Gdf11 and retinoids. Neuron 32, 997–1012 (2001).
Nordstrom, U., Maier, E., Jessell, T. M. & Edlund, T. An early role for WNT signaling in specifying neural patterns of Cdx and Hox gene expression and motor neuron subtype identity. PLoS Biol 4, e252 (2006).
Sharpe, C. & Goldstone, K. Retinoid signalling acts during the gastrula stages to promote primary neurogenesis. Int J Dev Biol 44, 463–470 (2000).
Sharpe, C. R. & Goldstone, K. Retinoid receptors promote primary neurogenesis in Xenopus. Development 124, 515–523 (1997).
Pierani, A., Brenner-Morton, S., Chiang, C. & Jessell, T. M. A sonic hedgehog-independent, retinoid-activated pathway of neurogenesis in the ventral spinal cord. Cell 97, 903–915 (1999).
Lippmann, E. S., Al-Ahmad, A., Palecek, S. P. & Shusta, E. V. Modeling the blood-brain barrier using stem cell sources. Fluids Barriers CNS 10, 2 (2013).
Winkler, E. A., Bell, R. D. & Zlokovic, B. V. Central nervous system pericytes in health and disease. Nat Neurosci 14, 1398–1405 (2011).
Crisan, M. et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell 3, 301–313 (2008).
Kusuma, S. et al. Self-organized vascular networks from human pluripotent stem cells in a synthetic matrix. Proc Natl Acad Sci U S A 110, 12601–12606 (2013).
Wright, L. S. et al. Gene expression in human neural stem cells: effects of leukemia inhibitory factor. J Neurochem 86, 179–195 (2003).
Crone, C. & Olesen, S. P. Electrical resistance of brain microvascular endothelium. Brain Res 241, 49–55 (1982).
Smith, Q. R. & Rapoport, S. I. Cerebrovascular permeability coefficients to sodium, potassium and chloride. J Neurochem 46, 1732–1742 (1986).
Acknowledgements
We would like to thank the WiCell Research Institute for providing reagents and Dr. Clive Svendsen (Cedars-Sinai Medical Center, Los Angeles, CA) for providing the human NPCs used in this study7. E.S.L. was supported by a Chemistry Biology Interface training fellowship (T32 GM008505) and S.M.A. was supported by a National Science Foundation graduate fellowship. This work was funded in part by the US National Institutes of Health (NIH) grants AA020476 and NS083688 to E.V.S. and S.P.P.
Author information
Authors and Affiliations
Contributions
E.S.L., A.A.-A., S.M.A., S.P.P. and E.V.S. designed research. E.S.L., A.A-A. and S.M.A. performed experiments. E.S.L., A.A-A., S.M.A., S.P.P. and E.V.S. analyzed data and wrote the paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Lippmann, E., Al-Ahmad, A., Azarin, S. et al. A retinoic acid-enhanced, multicellular human blood-brain barrier model derived from stem cell sources. Sci Rep 4, 4160 (2014). https://doi.org/10.1038/srep04160
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep04160
This article is cited by
-
Mouse embryonic stem cell-derived blood–brain barrier model: applicability to studying antibody triggered receptor mediated transcytosis
Fluids and Barriers of the CNS (2023)
-
The maturation of iPS cell-derived brain microvascular endothelial cells by inducible-SOX18 expression
Fluids and Barriers of the CNS (2023)
-
SARS-CoV-2 infects epithelial cells of the blood-cerebrospinal fluid barrier rather than endothelial cells or pericytes of the blood-brain barrier
Fluids and Barriers of the CNS (2023)
-
Characterization of an iPSC-based barrier model for blood-brain barrier investigations using the SBAD0201 stem cell line
Fluids and Barriers of the CNS (2023)
-
Human isogenic cells of the neurovascular unit exert transcriptomic cell type-specific effects on a blood-brain barrier in vitro model of late-onset Alzheimer disease
Fluids and Barriers of the CNS (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
