
Abstract
Applying two-dimensional monolayer materials in nanoelectronics and spintronics is hindered by a lack of ordered and separately distributed spin structures. We investigate the electronic and magnetic properties of one-dimensional zigzag and armchair 3d transition metal (TM) nanowires on graphyne (GY), using density functional theory plus Hubbard U (DFT + U). The 3d TM nanowires are formed on graphyne (GY) surfaces. TM atoms separately and regularly embed within GY, achieving long-range magnetic spin ordering. TM exchange coupling of the zigzag and armchair nanowires is mediated by sp-hybridized carbon and results in long-range magnetic order and magnetic anisotropy. The magnetic coupling mechanism is explained by competition between through-bond and through-space interactions derived from superexchange. These results aid the realization of GY in spintronics.
Similar content being viewed by others
Introduction
Two-dimensional (2D) monolayer materials have attracted much interest, because of their attractive properties and potential in nanoscale devices1,2,3,4,5. Manipulating the magnetic structures of 2D monolayer sheets such as graphene and h-BN through transition metal (TM) adsorption is an important challenge for realizing their application in spintronics and nanoelectronics6,7,8. The clustering of TM adatoms on such materials is energetically favorable9, because of the weak binding between TM adatoms and graphene or h-BN10,11,12,13,14,15. Obtaining stable spin ordering by TM adatoms is therefore difficult on these materials, which hampers their applications in spintronics. Developing new magnetic structures with ordered spin arrangements is highly desirable for such applications. Zhou et al.16 embedded TM atoms in 2D phthalocyanine sheets, achieving separate regularly distributed TM atoms within a monolayer. Graphyne (GY) consisting of hexagonal carbon rings and acetylene linkages is a 2D carbon allotrope of similar symmetry to graphene and its synthesis has been theoretically predicted17,18. Much effort has been devoted to its synthesis and numerous related polymeric structures have been reported19,20. Graphdiyne (GDY) is closely related to GY and was recently prepared on a copper surface21,22. GY is energetically more stable than GDY, so its future preparation is expected. We previously found that a single adsorbed 3d TM atom on GY (TM-GY) promotes the magnetization of the system, which suggests its potential in spintronics23. The additional px − pyπ/π* state of sp-hybridized carbon in GY allows it to strongly bind the TM adatom and easily trap it within its alkyne rings23. In contrast to clustered TM adatoms on graphene and h-BN, dispersed adsorbed TM adatoms are energetically favorable on GY. GY may be a potential template for synthesizing ordered spin arrangements for future spintronics. Long-range indirect exchange coupling between magnetic adatoms mediated by host atoms is important for achieving magnetic ordering. For example, Ruderman-Kittel-Kasuya-Yoshida exchange coupling occurs between two magnetic impurities in graphene, which originates from its semi-metallic nature24,25. Understanding the magnetic alignment of TM adatoms on GY, especially their magnetic coupling mechanism mediated by sp-hybridized carbon, is important for applying TM-GY in spintronics.
One-dimensional (1D) nanoscale magnetism with ferromagnetic (FM) spin order is desirable in nanoelectronics and spintronics. Spin polarized 1D nanostructures such as graphene nanoribbons26 and organometallic nanowires27,28 show strong potential in spintronics. Dimensionally reduced TM structures such as dimers29 and chains30,31,32 have been shown to lead to large magnetic anisotropy. Magnetic anisotropy in TM nanowires leads to interesting magnetic properties, including ballistic anomalous magnetoresistance33 and spin and magnetization tunneling34.
In view of the strong binding between TM adatoms and GY, we construct 1D ordered TM nanowires on GY and investigate their stability, magnetic ordering and magnetic anisotropy. Advances in STM tip manipulation35,36,37 suggest that TM nanowires on GY and related surfaces may be experimentally achieved. The current results show that exchange coupling of TM nanowires mediated by sp-hybridized carbon gives rise to long-range magnetic ordering. Fe and Co nanowires exhibit relatively large magnetic anisotropy energies (MAE) and orbital magnetic moments. These results aid the realization of GY-related materials in spintronics.
Results
Geometry and stability of adsorbed TM nanowires
Before investigating the magnetic properties of TM nanowires adsorbed on GY (TM-chain-GY), we consider the stability of TM dimers adsorbed on GY (TM-dimer-GY). We first investigate the most stable TM (V, Cr, Mn, Fe and Co) dimer configurations on the GY surface. The most stable adsorption site for a single TM atom is at the alkyne ring of GY23. Based on this configuration, the second TM is located on the surface. Two possible initial adsorption sites for the second TM are considered, as shown in Fig. 1. The H1 site is at the neighboring hexagonal ring of the first TM atom. The H2 site is at the neighboring alkyne ring of the first TM atom. CON2 is the most stable configuration, as shown in Fig. 1. This configuration involves both TM atoms trapped in the neighboring alkyne rings by -C≡C- bonds. The CON1 configuration involves one TM trapped in the alkyne ring and another adsorbed at the hollow site of the hexagonal ring. The CON1 configuration energies are 1.097, 0.515, 0.573, 0.978 and 1.084 eV per TM atom less stable than those of the CON2 configuration, for V, Cr, Mn, Fe and Co, respectively. All five TM dimers are chemisorbed on GY. The configurations have large adsorption energies of −2.780, −1.256, −0.942, −3.096 and −2.200 eV per TM atom for the V, Cr, Mn, Fe and Co dimers, respectively. The adsorption energies per TM adatom in TM-dimer-GY are similar to those of single TM adatoms on GY. Adsorption energies for single TM adatoms on GY are −1.672, −1.331, −1.433, −2.424 and −2.541 eV for V, Cr, Mn, Fe and Co, respectively. The V and Fe dimers are more stable than their corresponding single adsorbed TM atoms. Stability is derived from the -C≡C- bonding. The px and py orbitals of sp-hybridized carbon in GY contribute to σ- and π-bonding, which facilitates bonding with the TM adatom. Increasing the number of TM atoms yields adsorption energies similar to those for the dimer configuration. This suggests that regular TM nanowires could be constructed on GY surfaces.

Schematic of the (upper) ZTMW and (lower) ATMW unit cells.
The CON1 and CON2 configurations of TM dimers are also shown (lower). Cartesian and polar coordinates are used to define the magnetization direction (z′). θ and ϕ represent the azimuthal and polar angles.
After obtaining stable TM-dimer-GY configurations, we now consider TM-chain-GY. Zigzag transition metal nanowires (ZTMW) and armchair transition metal nanowires (ATMW) are considered as two TM-chain-GY types and are shown in Fig. 1. Adsorption energies and structural parameters of equilibrium ZTMW and ATMW configurations are listed in Tab. I. Table I shows that ZTMW and ATMW nanowires generally exhibit similar geometries and adsorption energies. Therefore, the discussion is largely focused on the ZTMW system. The considerable negative adsorption energies for all of the 3d ZTMW and ATMW nanowire systems indicate chemisorption. The weakest chemisorption occurs in ZMnW, whose adsorption energy is −0.907 eV per TM atom. The strongest chemisorption occurs in ZFeW, whose adsorption energy is −2.914 eV per TM atom. Table I shows that TM atoms in ZTMW and ATMW exhibit similar stabilities. Stability in all cases originates from carbon within -C≡C- bonding, as reported previously23. Valence electrons of the TM atom couple with the pz and px−y orbitals of carbon, in the adsorbed TM nanowires. Table I indicates that the maximum and minimum distances between the TM adatom and nearest carbon atom (dAC) in ZTMW are 2.29 and 2.03 Å for V and Mn, respectively. All TM adatoms of the TM chains protrude slightly from the GY plane, except for Mn. The largest height (h) is 1.47 Å for V and the smallest h is 0 Å for Mn. The size of the Mn adatom is well-suited to that of the acetylene ring of GY.
Magnetic ordering and electronic structure of TM nanowires on GY
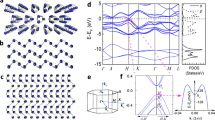
Magnetic coupling is investigated based on the most stable adsorbed TM dimer and nanowire configurations. Distinct spin polarization is found for GY with adsorbed TM dimers and nanowires. Both collinear FM and antiferromagnetic (AFM) coupling are considered for all TM-dimer-GY and TM-chain-GY configurations. The energy difference ΔE = EAFM − EFM between the FM and AFM states per unit cell is shown in Tables I and II, for the TM-nanowire-GY and TM-dimer-GY systems, respectively. Positive and negative energy differences between the FM and AFM states indicate FM and AFM ground states, respectively. The ground states for the V, Mn, Fe and Co dimers are FM, whereas that for the Cr dimer is AFM. ΔE for the Mn dimer is larger than 100 meV, which is necessary for high temperature FM systems. In ZTMW and ATMW, TM adatoms for V, Mn, Fe and Co exhibit FM coupling characteristics, whereas Cr exhibits AFM coupling with a zero magnetic moment. The large ΔE for ZVW, ZMnW and AMnW should stabilize their FM nature at high temperature. The band structures for the ZTMW systems are shown in Fig. 2. It is evident that the adsorption of TM nanowires on GY modulates its electronic properties and that all ZTMW systems exhibit metallic characteristics. The ZTMW systems all exhibit similar band structures, as derived from their quais-1D electronic structure. This is similar to the behavior of carbon nanotubes38, graphene nanoribbons39 and Cr nanowires embedded in topological defects in graphene40. Similar to ZTMW, the ATMW systems also exhibit metallic characteristics. We previously reported that GY magnetization emerged upon the adsorption of a single TM atom23. However, magnetic ordering in TM nanowires arising from magnetic exchange coupling between TM atoms deserves a thorough investigation. Table I indicates that the FM states of all non-Cr systems are energetically more favorable than the AFM states. ZCrW and ACrW exhibit AFM coupling with zero net magnetic moments. ZCrW and ACrW exhibit −42 and −93 meV differences between the FM and AFM states, respectively. TM nanowires on GY surfaces exhibit robust FM or AFM ordering, indicating their suitability for spintronics. ZFeW, ZCrW, AFeW and ACrW are used as examples to visualize spin ordering in ZTMW and ATMW. The spin charge densities (SCD) of these systems are shown in Fig. 3. The magnetic moment of TM-chain-GY is largely distributed around the Fe and Cr atoms. The sp-hybridized carbon carries a very small magnetic moment, because of the hybridization between the TM and carbon. The SCD results indicate that the spin polarized charge density is largely localized within the TM chain region. This suggests that coupling between neighboring chains is weak. This is crucial for achieving controlled TM nanowire spin-polarized currents for spintronics.

Band structures of (a) ZVW, (c) ZCrW, (d) ZMnW, (e) ZFeW and (f) ZCoW in the FM state and (b) ZCrW in the AFM state.
Dashed red and solid green lines represent the majority and minority spin channels, respectively. The Fermi level is set to zero.

SCD distributions for (a) AFeW, (b) ZFeW, (c) ACrW and (d) ZCrW on GY.
Light green and dark blue isosurfaces denote spin-up and spin-down charge densities, respectively. The isosurface charge density is 0.004e/Å3.
To understand the origin of magnetism in the TM nanowires, partial densities of states (PDOS) of TM atoms and their nearest neighbor (NN) carbon atoms are shown in Fig. 4. The results are shown for ZTMW and those of ATMW are similar. The PDOS of ATMW are shown in Supplementary Fig. S1. The PDOS of only one NN sp-hybridized carbon is shown, because each NN carbon exhibits similar characteristics. The magnetic moments of Cr, Mn, Fe and Co in the dimers and nanowires are all lower than in their freestanding states, except for that of V. A reduced magnetic moment originates from electron transfer from the TM to GY and also from electron redistribution of the TM d state which is induced by the strong coupling between the TM and GY. This is similar to our previous report for single TM adatoms on GY23. ZFeW is used as an example, to explain the origin of magnetism in TM-dimer-GY and TM-nanowire-GY. The PDOS of the Fe adatom and its NN carbon atoms for ZFeW are shown in Fig. 4(e). The TM absorbed on GY possesses C3V symmetry, in which the five d orbitals are generally split into  , E1 (dyz, dxz) and E2 (dxy,
, E1 (dyz, dxz) and E2 (dxy,  ). For the majority spin, the Fe d orbital strongly couples with the in-plane px–y and out-of-plane pz orbitals of its NN carbon atoms, in the range −6.4 ~ −2.1 eV. For the minority spin, there is overlap of the A1, E1 and E2 states of Fe and the px–y and pz states of the NN carbon atoms at −2.8 ~ −1.2 eV. The TM-chain-GY systems exhibit strong coupling between the TM d states and carbon out-of-plane pz and in-plane px–y states. Such hybridization lowers the energy of the Fe d states to below the Fermi level and induces electron redistribution within Fe. The Hubbard U also severely influences the energy of TM d orbitals, the details of which are shown in Supplementary Fig. S4. To evaluate charge transfer between the TM adatom and GY, an atomic basin charge based on the atoms in molecular (AIM) theory41 is adopted. Charge transfer between the TM and GY is qualitatively determined from comparing the valence electrons in the TM atomic basin of the TM nanowire system, with those of corresponding free standing state. Details of the electron transfer from the TM adatom to GY (Te) are shown in Tab. I. There is considerable electron exchange between the TM and GY. This originates from the strong coupling between the TM d states and NN carbon p states, which determines the magnetic moment of TM nanowire.
). For the majority spin, the Fe d orbital strongly couples with the in-plane px–y and out-of-plane pz orbitals of its NN carbon atoms, in the range −6.4 ~ −2.1 eV. For the minority spin, there is overlap of the A1, E1 and E2 states of Fe and the px–y and pz states of the NN carbon atoms at −2.8 ~ −1.2 eV. The TM-chain-GY systems exhibit strong coupling between the TM d states and carbon out-of-plane pz and in-plane px–y states. Such hybridization lowers the energy of the Fe d states to below the Fermi level and induces electron redistribution within Fe. The Hubbard U also severely influences the energy of TM d orbitals, the details of which are shown in Supplementary Fig. S4. To evaluate charge transfer between the TM adatom and GY, an atomic basin charge based on the atoms in molecular (AIM) theory41 is adopted. Charge transfer between the TM and GY is qualitatively determined from comparing the valence electrons in the TM atomic basin of the TM nanowire system, with those of corresponding free standing state. Details of the electron transfer from the TM adatom to GY (Te) are shown in Tab. I. There is considerable electron exchange between the TM and GY. This originates from the strong coupling between the TM d states and NN carbon p states, which determines the magnetic moment of TM nanowire.

PDOS for (a) V, (c) Cr, (d) Mn, (e) Fe and (f) Co in the FM state and (b) Cr in the AFM state.
Positive and negative lines represent the majority and minority spin channels, respectively. The PDOS of the (upper) NN carbon and (lower) TM adatom, respectively. The PDOS of only one of the two TM atoms in the unit cell is shown. The PDOS of the adjacent Cr in (b) is positive. The Fermi level is set to zero.
We now discuss the origin of the magnetic ordering of TM nanowires on GY. TM-chain-GY consists of weakly interacting TM atoms. Their magnetic coupling follows the competition between through-bond and through-space coupling, described by the superexchange mechanism. This mechanism has been used to explain the origin of cation vacancies in GaN42, 2D TM dichalcogenides43 and 1D organometallic wires28. The spin exchange parameter J for superexchange can be divided into FM and AFM components, as determined by the overlap integral and overlap density, respectively. The through-bond (TM-C-TM indirect) and through-space (TM-TM direct) interactions qualitatively originate from the FM and AFM components, respectively. Mechanistic details are available elsewhere44,45. The through-space interaction is favored when the interaction of TM atoms produces a large energy split. The through-bond spin polarization can be considered as an indirect interaction mediated by ligand atoms. The magnetic coupling of TM nanowires on GY is mediated by the sp-hybridized carbon. Thus, the magnetic coupling can be determined by the competition between the through-bond and through-space coupling interactions. In through-bond coupling, the TM atom with spin up (spin down) polarization will induce spin down (spin up) polarization of its directly bound carbons and will further induce spin up (spin down) polarization of its NN TM, forming the spin arrangement[↑↓↑↓]n. This is shown in Fig. 5(a). In through-space coupling, the TM atom with spin up polarization will induce spin down polarization of its NN TM, forming the spin arrangement [↑ − ↓ − ↑ − ↓]n, as shown in Fig. 5(b). Fe and Cr are used as examples, to elucidate the origin of magnetic ordering in TM nanowires adsorbed on GY. For ZFeW and AFeW, there is a localized magnetic moment for the NN carbons of Fe, whose polarization is AFM with respect to that of the directly bound Fe. The PDOS are shown in Fig. 4(e) and Supplementary Fig. S1(e) and indicate that the NN sp-hybridized carbons of Fe exhibit spin splitting. Through-bond spin polarization occurs along the TM-C-TM path, by mediation of the p orbital of the NN sp-hybridized carbons, forming the spin arrangement [↑↓↑↓]n. Therefore, all Fe atoms in AFeW and ZFeW exhibit FM coupling and maintain long-range FM spin alignment, as shown in Fig. 3(a) and (b). The PDOS in Fig. 4(e) and Supplementary Fig. S1(e) indicate that although the bonding of these systems is largely dictated by the E1 and E2 states, their anti-bonding states admix with A1 states. Such admixing enhances the through-space interaction, which reduces the energy difference between the FM and AFM states. Similar results are exhibited for ZCoW and ACoW, producing smaller ΔE values. For ZVW, AVW, ZMnW and AMnW, bonding and anti-bonding states largely originate from the E1 and E2 states, resulting in higher ΔE values. In ACrW and ZCrW, carbon atoms surrounding Cr exhibit largely symmetric majority and minority states, as shown in Fig. 4(b) and Supplementary Fig. S1(b). AFM coupling of Cr in ACrW and ZCrW follows the through-space interaction, in agreement with SCD results in Fig. 3. Spin up polarized Cr induces spin down polarization on its nearest neighbor Cr, forming the spin arrangement [↑ − ↓ − ↑ − ↓]n. The PDOS shown in Fig. 4(b) and Supplementary Fig. S1(b) indicates that the A1 states strongly admix in their bonding states, which enhances the through-space coupling and produces AFM ordering. In summary, the magnetic coupling of TM nanowires adsorbed on GY occurs via competition between through-bond and through-space coupling, originating from the superexchange mechanism. Through-bond coupling dominates in the FM coupling between TM atoms. Through-space coupling dominates in ACrW and ZCrW, producing AFM ordering. This is because the A1 state admixes in the bonding state.

Schematic showing (a) through-bond and (b) through-space spin polarizations.
Magnetic anisotropy of TM nanowires on GY
The reduced dimensionality of TM nanowires can enhance the magnetic anisotropy and produce interesting properties. Previous studies31,32,46 indicate that 1D chains or wires on metal surfaces show larger MAE values than those of their bulk 3D materials. Investigating magnetic anisotropy in 1D TM nanowires on GY is of interest. Whether the easy axis is parallel or perpendicular to the GY plane is an important question.
We performed noncollinear magnetic calculations using the GGA (PBE) exchange-correlation functional for ZTMW and ATMW. The MAE is induced by spin-orbit coupling (SOC), so SOC is included in the MAE calculations. The spin magnetic moment under the easy axis of magnetization and the orbital magnetic moment under the easy and hard axes of magnetization were calculated for the TM chain. The results are shown in Tab. III. For the easy axis of magnetization, the sequence of orbital magnetic moments of TM adatoms is Co > Fe > V > Mn > Cr, for both ZTMW and ATMW. ZFeW, AFeW, ZCoW and ACoW exhibit considerable orbital magnetic moments of 0.112, 0.103, 0.230 and 0.153 μB, respectively. ZTMW is used as an example, to characterize the MAE of the TM nanowire systems. The total energy while rotating the magnetization orientation (magnetization orientation is defined by the azimuth (θ) and polar (ϕ) angles with respect to the z and x axes, respectively, as shown in Fig. 1) is calculated (i) from the x (90,0) to z (0,0) axes within the xz plane, (ii) from the y (90,90) to z (0,90) axes within the yz plane and (iii) from the x (90,0) to y (90,90) axes within the xy plane, by 10 degree intervals using the MAE per unit cell definition as follows:



where E(θ, ϕ) is the total energy per unit cell, when the magnetization orientation is (θ, ϕ). Figure 6 shows MAExz, MAEyz and MAExy as a function of rotation angle, for all ZTMW systems. ZFeW and ZCoW exhibit easy-axis anisotropy and easy-plane anisotropy with relatively large MAE values of 2.422 and −2.076 meV, respectively, as shown in Fig. 6. The ATMW systems exhibit similar results. The MAE values of the Co and Fe nanowire systems are similar to that of a Co chain on Pt(111)30, Co dimer on graphene and Fe doped carbon nanotubes46,47,48. Such MAE values cannot compare with those of 4d and 5d TM-doped systems29, but are considerable for 3d TM-doped systems. ZVW, AVW, ZCrW, ACrW, ZMnW and AMnW exhibit easy-axis anisotropy with MAE values of 0.816, 1.028, 0.605, 0.906, 1.054 and 1.121 meV, respectively. Figure 6 shows that the MAE is small, except for that of ZVW with in-plane anisotropy and a MAE of 0.522 meV, when the quantization axis lies in the xy plane.

MAE as a function of magnetization angle from the hard to easy axis for ZTMW.
[x-z] represents from the x to z, [y-z] from the y to z and [x-y] from the x to y axes.
ZTMW is used as an example to study the origin of the magnetic anisotropy. Magnetic anisotropy is derived from the SOC of the system. The spin-conserving term of the SOC Hamilton  is defined as45,49
is defined as45,49

where λ is the SOC constant and θ is the azimuthal angle of the magnetization direction with the coordinates of TM-GY (shown in Fig. 1). By treating the SOC Hamilton as second perturbations, the occupied (Ψo) and unoccupied (Ψu) states can interact via the matrix element 〈 〉. The associated energy lowering can be expressed in simplified form as:
〉. The associated energy lowering can be expressed in simplified form as:

The interaction between occupied and unoccupied states determines the preferred spin orientation. d states with the same |m| value interact through the operator  to give the non-zero matrix element. Different |m| values interact through the ladder operators
to give the non-zero matrix element. Different |m| values interact through the ladder operators  and
and  45,49.
45,49.
V nanowires on GY exhibit easy-axis anisotropy. The results shown in Fig. 7(a) indicate that the HOMO and LUMO of V nanowires belong to the same spin channel (majority) and that they are largely derived from the |m| = 1(dyz,dxz) and |m| = 2( , dxy) states. The energy lowering associated with the HOMO-LUMO interaction induced by SOC depends on the matrix element
, dxy) states. The energy lowering associated with the HOMO-LUMO interaction induced by SOC depends on the matrix element

From equation 4, the nonzero matrix element, depending on the magnetization direction, can be obtained as

When θ = 0° (easy-axis anisotropy), the energy is lowered to a minimum and V d states with |m| = 1 interact with each other through  . This is in agreement with the easy-axis anisotropy for ZVW. For Cr nanowires, the HOMO and LUMO states for AFM and FM coupling largely originate from the |ml| = 2 states, as shown in Fig. 7(b) and (c). Equations 4 and 5 suggest that
. This is in agreement with the easy-axis anisotropy for ZVW. For Cr nanowires, the HOMO and LUMO states for AFM and FM coupling largely originate from the |ml| = 2 states, as shown in Fig. 7(b) and (c). Equations 4 and 5 suggest that  couples the occupied and unoccupied states of
couples the occupied and unoccupied states of  and dxy. Coupling lowers the energy of the perpendicular magnetization direction and the system exhibits easy-axis anisotropy. Figure 7(d) shows that the interaction between the HOMO and LUMO is largely contributed by the
and dxy. Coupling lowers the energy of the perpendicular magnetization direction and the system exhibits easy-axis anisotropy. Figure 7(d) shows that the interaction between the HOMO and LUMO is largely contributed by the  and dxy states of minority spins for ZMnW. The interaction between
and dxy states of minority spins for ZMnW. The interaction between  and dxy through
and dxy through  leads to the easy-axis anisotropy. The |ml|-resolved PDOS of ZCoW are shown in Fig. 7(e). The HOMO largely originates from the |ml| = 1 and |ml| = 2 states, whereas the LUMO largely originates from the |ml| = 1 state. SOC induces the interaction between |ml| = 1 of the HOMO and LUMO by
leads to the easy-axis anisotropy. The |ml|-resolved PDOS of ZCoW are shown in Fig. 7(e). The HOMO largely originates from the |ml| = 1 and |ml| = 2 states, whereas the LUMO largely originates from the |ml| = 1 state. SOC induces the interaction between |ml| = 1 of the HOMO and LUMO by  , so the system exhibits easy-axis anisotropy. The interaction between the HOMO |ml| = 2 and LUMO |ml| = 1 by
, so the system exhibits easy-axis anisotropy. The interaction between the HOMO |ml| = 2 and LUMO |ml| = 1 by  favors easy-plane anisotropy. The easy-axis anisotropy for Fe-chain-GY originates from competition between
favors easy-plane anisotropy. The easy-axis anisotropy for Fe-chain-GY originates from competition between  and
and  . The |ml|-resolved PDOS of ZCoW are shown in Fig. 7(f) and indicate that the HOMO and LUMO are largely contributed by the |ml| = 2 and |ml| = 1 states. These states interact through
. The |ml|-resolved PDOS of ZCoW are shown in Fig. 7(f) and indicate that the HOMO and LUMO are largely contributed by the |ml| = 2 and |ml| = 1 states. These states interact through  and
and  . The energy-lowering associated with
. The energy-lowering associated with  approaches a maximum when θ = 90°. In summary, the magnetization direction for ZCoW is parallel to the plane, indicating easy-plane anisotropy.
approaches a maximum when θ = 90°. In summary, the magnetization direction for ZCoW is parallel to the plane, indicating easy-plane anisotropy.

Majority (red) and minority (blue) spin ml-resolved PDOS for (a) V, (c) Cr, (d) Mn, (e) Fe and (f) Co in the FM state and (b) Cr in the AFM state.
The Fermi level is set to zero.
Discussion
We have performed DFT + U calculations on the magnetic exchange coupling and anisotropy of 3d TM zigzag and armchair nanowires adsorbed on GY. TM magnetic coupling mediated by sp-hybridized carbon results in a robust long-range magnetic ordering of the nanowires. Magnetic coupling is explained by competition between through-bond and through-space interactions, derived from the superexchange mechanism. Fe and Co nanowires exhibit considerable MAE values and orbital magnetic moments. V, Cr, Mn and Fe nanowires exhibit easy-axis anisotropy and Co nanowires exhibit easy-plane anisotropy. The in-plane anisotropy energy is close to zero, except for in V nanowires.
To investigate the dependence of magnetic properties on the width of the TM nanowire system, we enlarged the configurations to double zigzag and double armchair TM nanowires. The TM nanowires form two types of graphene-like honeycomb nanoribbons: zigzag TM nanoribbons (ZTMNs) and armchair TM nanoribbons (ATMNs). These are shown in Supplementary Fig. S2 and are similar to zigzag and armchair graphene nanoribbons. We investigated the magnetic order of the ZTMNs and ATMNs configurations. The energy differences between the FM and AFM states of the interwire and intrawire coupling are defined as ΔEinter = EAFM1 − EFM and ΔEinter = EAFM2 − EFM, respectively. EFM, EAFM1 and EAFM2 represent the energy of FM coupling, interwire AFM coupling and intrawire AFM coupling, respectively. The results are shown in Supplementary Table S1. For V, Mn, Fe and Co, the ZTMNs and ATMNs exhibit FM coupling characteristics, similar to those of ZTMW and ATMW. The ground states of ZCrNs and ACrNs exhibit intrawire AFM coupling, with zero magnetic moments. Intrawire coupling is generally stronger than interwire coupling. The dependence of magnetic anisotropy on nanowire width is also presented. Within the size investigated in the current study, the dependence of the magnetic anisotropy on nanowire size is slight. Only the results of ZFeNs and ZCoNs are presented, because the magnetic anisotropy for the other systems is small. Calculated MAE values and easy and hard axes for ZFeNs and ZCoNs are shown in Supplementary Table S2. MAE values for ZFeNs and ZCoNs are 2.038 and −2.378 meV, respectively. ZFeNs and ZCoNs exhibit easy-axis anisotropy and easy-plane anisotropy, respectively, similar to the results for the single TM nanowires.
This study has largely focused on the properties of TM nanowires on GY. The above discussion indicates that TM atoms can be dispersed on GY surfaces. Thus, TM adatoms can also form 2D configurations such as hexagonal rings. Calculated magnetic properties and adsorption energies of hexagonal TM rings on GY are shown in Supplementary Fig. S3 and Supplementary Table S3. Hexagonal TM rings on the GY surface exhibit similar magnetic coupling to that of nanowires. The adsorption energy per TM atom indicates that forming 2D clusters is as favorable as forming 1D nanowires. The magnetic and structural properties of 2D TM clusters require further investigation.
Method and computational details
For the single TM, TM dimer, ATMW and ZTMW systems, 2 × 2, 2 × 2, 2 × 3 and 2 × 3 super-cells were adopted, respectively. Such super-cells guaranteed that distances between TM atoms within adjacent images were larger than 13 Å. This was sufficient to avoid interactions between TM atoms in adjacent images. A vacuum space of 13 Å perpendicular to the GY plane was chosen to avoid interaction between neighboring images. First-principles calculations were performed using the Vienna ab initio simulation package (VASP)50,51 within spin-polarized DFT52,53. To account for the correlation energy of the localized 3d orbital of TM, the Hubbard U correction (DFT + U)54 was employed, based on the generalized gradient approximation of Perdew-Burke-Ernzerhof55. Projector augmented wave (PAW) potentials56,57 described the electron-ionic core interaction. A plane-wave basis set with a kinetic energy cutoff of 400 eV was employed. The Brillouin zone (BZ) was sampled using 5 × 3 × 1 and 9 × 5 × 1 gamma-centered Monkhorst-Pack grids, to calculate the relaxation and electronic structures, respectively. The criteria of energy and atom force convergence were 10−5 eV/unit cell and 0.01 eV/Å, respectively. The dipole correction58 was considered to deal with the varying potential distribution introduced by TM adsorption. All calculation parameters were systematically optimized. The rotationally invariant DFT + U formalism proposed by Dudarev et al. was used. Ueff = U − J was used instead of individual U and J values59. Wu et al. reported a study of TM-doped graphene systems, based on GGA + U. The Hubbard U of TM dimers could safely use that of single TM atoms, because of localization of the TM dopant60. We calculated the Hubbard U of the Fe-dimer and ZFeW, using linear-response theory. The Ueff of the Fe dimer and ZFeW were only 0.2 eV lower than that of a single Fe on GY. This deviation in Ueff does not influence the conclusions of our study. The linear-response method is very time-consuming, so we assigned Ueff = 4.84, 3.55, 5.01, 5.03 and 6.31 eV for V, Cr, Mn, Fe and Co adatoms, respectively, based on our earlier calculations23 using the linear response method61. SOC was included in the magnetic anisotropy calculations for self-consistency. The MAE was calculated from the ground state energies, for the magnetization axis aligned along the most relevant direction. Total energies were converged to a precision of 10−7 eV in MAE calculations.
To evaluate the stability of TM adatom configurations, the adsorption energy per TM atom (Ea) was adopted and defined as

where ETM+GY denotes the spin-polarized total energy of the adsorbed TM, EGY is the total energy of an isolated GY sheet and ETM is the spin-polarized total energy of the free TM atom.
To better understand the magnetic coupling of the TM-GY systems, SCD was used in all investigated systems and defined as

where  and
and  are the spin up and spin down charge densities of the TM-GY system, respectively.
are the spin up and spin down charge densities of the TM-GY system, respectively.
References
Novoselov, K. S. et al. Electric field effect in atomically thin carbon films. Science 306, 666–669 (2004).
Berger, C. et al. Functionalized Single Graphene Sheets Derived from Splitting Graphite Oxide. J. Phys. Chem. B 108, 19912–19916 (2004).
Fal'Ko, V. Graphene: Quantum information on chicken wire. Nat. Phys. 3, 151–152 (2007).
Son, Y. W., Cohen, M. L. & Louie, S. G. Half-metallic graphene nanoribbons. Nature 444, 347–349 (2006).
Wang, X. et al. N-doping of graphene through electrothermal reactions with ammonia. Science 324, 768–771 (2009).
Huang, B., Xiang, H., Yu, J. & Wei, S. H. Effective Control of the Charge and Magnetic States of Transition-Metal Atoms on Single-Layer Boron Nitride. Phys. Rev. Lett. 108, 206802 (2012).
Cocchi, C., Prezzi, D., Calzolari, A. & Molinari, E. Spin-transport selectivity upon Co adsorption on antiferromagnetic graphene nanoribbons. J. Chem. Phy. 133, 124703(2010).
Chan, K. T., Lee, H. & Cohen, M. L. Gated adatoms on graphene studied with first-principles calculations. Phys. Rev. B 83, 035405 (2011).
Porter, C. D. & Stroud, D. Clustering and magnetic anisotropy of Fe adatoms on graphene. Phys. Rev. B 85, 235452 (2012)
Johll, H., Kang, H. C. & Tok, E. S. Density functional theory study of Fe, Co and Ni adatoms and dimers adsorbed on graphene. Phys. Rev. B 79, 245416(2009).
Sevincli, H., Topsakal, M., Durgun, E. & Ciraci, S. Electronic and magnetic properties of 3d transition-metal atom adsorbed graphene and graphene nanoribbons. Phys. Rev. B 77, 195434 (2008).
Chan, K. T., Neaton, J. B. & Cohen, M. L. First-principles study of metal adatom adsorption on graphene. Phys. Rev. B 77, 235430(2008).
Cao, C., Wu, M., Jiang, J. & Cheng, H. P. Transition metal adatom and dimer adsorbed on graphene: Induced magnetization and electronic structures. Phys. Rev. B 81, 205424 (2011).
Li, J., Hu, M. L., Yu, Z., Zhong, J. X. & Sun, L. Z. Structural, electronic and magnetic properties of single transition-metal adsorbed BN sheet: A density functional study. Chem. Phys. Lett. 532, 40–46 (2012)
Wehling, T. O., Lichtenstein, A. I. & Katsnelson, M. I. Transition-metal adatoms on graphene: Influence of local Coulomb interactions on chemical bonding and magnetic moments. Phys. Rev. B 84, 235110 (2011).
Zhou, J. & Sun, Q. Magnetism of phthalocyanine-based organometallic single porous sheet. J. Am. Chem. Soc. 133, 15113–15119 (2011).
Baughman, R. H., Eckhardt, H. & Kertesz, M. Structure-property predictions for new planar forms of carbon: Layered phases containing sp 2 and sp atoms. J. Chem. Phys. 87, 6687 (1987).
Narita, N., Nagai, S., Suzuki, S. & Nakao, K. Optimized geometries and electronic structures of graphyne and its family. Phys. Rev. B 58, 11009 (1998).
Kehoe, J. M. et al. Carbon Networks Based on Dehydrobenzoannulenes. 3. Synthesis of Graphyne Substructures. Org. Lett. 2, 969–972 (2000).
Johnson, C. A. I. I., Lu, Y. & Haley, M. M. Carbon Networks Based on Benzocyclynes. 6. Synthesis of Graphyne Substructures via Directed Alkyne Metathesis. Org. Lett. 9, 3725–3728 (2007).
Li, G. et al. Architecture of graphdiyne nanoscale films. Chem. Commun. 46, 3256–3258 (2010).
Liu, H. B., Xu, J. L., Li, Y. J. & Li, Y. L. Aggregate Nanostructures of Organic Molecular Materials. Acc. Chem. Res. 43, 1496–1508 (2010).
He, J. et al. Magnetic Properties of Single Transition-Metal Atom Absorbed Graphdiyne and Graphyne Sheet from DFT + U Calculations. J. Phys. Chem. C 116, 26313–26321(2012).
Black-Schaffer, A. M. RKKY coupling in graphene. Phys. Rev. B 81, 205416(2010)
Dugaev, V. K., Litvinov, V. I. & Barnas, J. Exchange interaction of magnetic impurities in graphene. Phys. Rev. B 74, 224438(2006)
Sahin, H., Ataca, C. & Ciraci, S. Electronic and magnetic properties of graphane nanoribbons. Phys. Rev. B 81(20), 205417(2010).
Maslyuk, V. V. et al. Organometallic benzene-vanadium wire: A one-dimensional half-metallic ferromagnet. Phys. Rev. Lett. 97, 097201(2006).
Ma, Y., Dai, Y., Wei, W. & Huang, B. Engineering intriguing electronic and magnetic properties in novel one-dimensional staircase-like metallocene wires. J. Mater. Chem. C 1, 941–946(2013).
Hu & Wu Ruqian Giant magnetic anisotropy of transition-metal dimers on defected graphene. arXiv:1305.5978 (2013).
Dasa, T. R., Ignatiev, P. A. & Stepanyuk, V. S. Effect of the electric field on magnetic properties of linear chains on a Pt (111) surface. Phys. Rev. B 85, 205447(2012).
Mokrousov, Y., Bihlmayer, G., Heinze, S. & Blugel, S. Giant magnetocrystalline anisotropies of 4d transition-metal monowires. Phys. Rev. Lett. 96, 147201(2006).
Tung, J. C. & Guo, G. Y. Systematic ab initio study of the magnetic and electronic properties of all 3d transition metal linear and zigzag nanowires. Phys. Rev. B 76, 094413(2007).
Velev, J., Sabirianov, R. F., Jaswal, S. S. & Tsymbal, E. Y. Ballistic anisotropic magnetoresistance. Phys. Rev. Lett. 94, 127203(2005).
Wernsdorfer, W. & Sessoli, R. Quantum phase interference and parity effects in magnetic molecular clusters. Science 284, 133–135 (1999).
Nilius, N., Wallis, T. M. & Ho, W. Development of one-dimensional band structure in artificial gold chains. Science 297, 1853–1856 (2002).
Braun, K. F. & Rieder, K. H. Engineering electronic lifetimes in artificial atomic structures. Phys. Rev. Lett. 88, 096801(2002).
Khajetoorians, A. A., Wiebe, J., Chilian, B. & Wiesendanger, R. Realizing all-spin-based logic operations atom by atom. Science 332, 1062–1064 (2011).
Neto, A. C., Guinea, F., Peres, N. M. R., Novoselov, K. S. & Geim, A. K. The electronic properties of graphene. Rev. Mod. Phys. 81, 109–162 (2009).
Charlier, J. C., Blase, X. & Roche, S. Electronic and transport properties of nanotubes. Rev. Mod. Phys. 79, 677–732 (2007).
Yang, Y., Xiao, Y., Ren, W., Yan, X. H. & Pan, F. Half-metallic chromium-chain-embedded wire in graphene and carbon nanotubes. Phys. Rev. B 84, 195447 (2011).
Bader, R. F. W. Atoms in Molecules A Quantum Theory. M. Oxford: Clarendon Press 1990.
Jin, H., Dai, Y., Huang, B. & Whangbo, M. H. Ferromagnetism of undoped GaN mediated by through-bond spin polarization between nitrogen dangling bonds. Appl. Phys. Lett. 94, 162505 (2009)
Ma, Y. et al. Evidence of the Existence of Magnetism in Pristine VX2 Monolayers (X = S, Se) and Their Strain-Induced Tunable Magnetic Properties. ACS nano 6, 1695–1701(2012).
Hay, P. J., Thibeault, J. C. & Hoffmann, R. Orbital interactions in metal dimer complexes. J. Am. Chem. Soc. 97, 4884–4899 (1975).
Xiang, H., Lee, C., Koo, H. J., Gong, X. & Whangbo, M. H. Magnetic properties and energy-mapping analysis. Dalton Trans. 42, 823–853(2013).
Blonski, P. & Hafner, J. Magnetic anisotropy of transition-metal dimers: Density functional calculations. Phys. Rev. B 79, 224418(2009).
Zou, X. et al. Preparing spin-polarized scanning tunneling microscope probes on capped carbon nanotubes by Fe doping: A first-principles study. Appl. Phys. Lett. 94, 193106 (2009).
Xiao, R. et al. Co dimers on hexagonal carbon rings proposed as subnanometer magnetic storage bits. Phys. Rev. Lett. 103, 187201(2009).
Dai, D., Xiang, H. & Whangbo, M. H. Effects of spin orbit coupling on magnetic properties of discrete and extended magnetic systems. J. Comput. Chem. 29, 2187–2209(2008).
Kresse, G. & Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169(1996).
Kresse, G. & Furthmuller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Matter. Sci. 6, 15–50 (1996).
Hohenberg, P. & Kohn, W. Inhomogeneous electron gas. Phys. Rev. 136, B864–B871 (1964).
Kohn, W. & Sham, L. J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 140, A1133–A11389 (1965).
Anisimov, V. I., Aryasetiawan, F. & Lichtenstein, A. I. First-principles calculations of the electronic structure and spectra of strongly correlated systems: the LDA + U method. J. Phys.: Condens. Matter 9, 7679(1997).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Blochl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953(1994).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 17589(1999).
Neugebauer, J. & Scheffler, M. Adsorbate-substrate and adsorbate-adsorbate interactions of Na and K adlayers on Al (111). Phys. Rev. B 46, 16067 (1992).
Dudarev, S. L., Botton, G. A., Savrasov, S. Y., Humphreys, C. J. & Sutton, A. P. (1998). Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA + U study. Phys. Rev. B 57, 1505 (1998).
Wu, M., Cao, C. & Jiang, J. Z. Electronic structure of substitutionally Mn-doped graphene. New J. Phys. 12, 063020(2010).
Cococcioni, M. & de Gironcoli, S. Linear response approach to the calculation of the effective interaction parameters in the LDA + U method. Phys. Rev. B 71, 035105 (2005).
Acknowledgements
This work is supported by the Program for New Century Excellent Talents in University (Grant No. NCET-10-0169), the National Natural Science Foundation of China (Grant Nos. 11032010 and 11274262), the Hunan Provincial Innovation Foundation for Postgraduate (Grant Nos. CX2012B273, CX2013B263).
Author information
Authors and Affiliations
Contributions
L.Z.S. and J.H. conceived the initial idea of this research. J.H. demonstrated the initial idea and collected all data. P.Z., N.J., S.Y.M., K.W.Z., R.Z.W. and L.Z.S. participated in the discussions and analyzed the data. J.H. and L.Z.S. wrote the manuscript. L.Z.S. designed and coordinated the project.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary information for "Magnetic Exchange Coupling and Anisotropy of 3d Transition Metal Nanowires on Graphyne"
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
He, J., Zhou, P., Jiao, N. et al. Magnetic Exchange Coupling and Anisotropy of 3d Transition Metal Nanowires on Graphyne. Sci Rep 4, 4014 (2014). https://doi.org/10.1038/srep04014
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep04014
This article is cited by
-
Graphdiyne: from Preparation to Biomedical Applications
Chemical Research in Chinese Universities (2021)
-
Diffusion Kinetics Study of Lithium Ion in the Graphdiyne Based Electrode
Chemical Research in Chinese Universities (2021)
-
Study of carrier mobility of tubular and planar graphdiyne
Applied Physics A (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
