
Abstract
Single-layered graphene has unique electronic, chemical and electromechanical properties. Recently, graphite exfoliation in N-methylpyrrolidone and molten salt has been demonstrated to generate monolayer exfoliated graphene sheets (EGS). However, these solvents are either high-priced or require special care and have high boiling points and viscosities, making it difficult to deposit the dispersed graphene onto substrates. Here we show a universal principle for the exfoliation of graphite in water to single-layered and several-layered graphene sheets via the direct exfoliation of highly oriented pyrolytic graphite (HOPG) using pyridinium tribromide (Py+Br3−). Electrical conductivity >5100 S/cm was observed for filtered graphene paper and the EGS exhibited superior performance as a hole transport layer compared to the conventional material N,N-di(naphthalene-1-yl)-N,N-diphenylbenzidine at low voltage. The overall results demonstrate that this method is a scalable process for the preparation of highly conductive graphene for use in the commercial manufacture of high-performance electronic devices.
Similar content being viewed by others

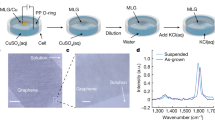

Introduction
Graphene, a novel two-dimensional and single-layer nanomaterial, has opened a new horizons in both fundamental science and advanced technology due to its unique electronic, chemical and electromechanical properties1,2,3,4,5. Because of these sensational properties, several methods have been established to prepare graphene such as bottom-up growth6,7,8,9,10 and top-down exfoliation5,11,12,13,14. According to the increasing demand of the large amount of graphene production15, the latter top-down approach is an appealing method from the point of view of chemist for the following reasons. First, it is simple, direct, facile producing graphene sheets by solvent treatment of graphite. Second, the exfoliated graphene sheets (EGS) form colloidal suspension in the solvents, thereby allowing their manipulation into various fabrication processes, such as spin-coating, mixing, chemical functionalization. Recently, a significant breakthrough was achieved when two autonomous groups demonstrated that graphite could be exfoliated in particular solvent to generate monolayer graphene12,16. The parpared graphene relies on using certain solvents, such as N-methyl-pyrrolidone or molten salt. However, these processes are not without its drawbacks. These solvents are either high-priced or need special care. Moreover, they have high boiling point and higher viscosity, making it hard to deposit on surfaces. In contrast, if the exfoliated graphene could be dispersed in a convenient medium, such as water, it could pave the way for many practical application developments. Based on previously published studies, the exfoliation of graphite to defect-free and unoxidized graphene sheets in aqueous medium is urgently required and still an important issue.
Results
The effect of four solvents, i.e., benzene, tetrabutylammonium perchlorate (TBAP), pyridinium tribromide (Py+Br3−) and 1-butyl-3-methylimidazolium tetrafluoroborate (BMI+BF4−) on the graphite exfoliation of highly oriented pyrolytic graphite (HOPG) was investigated using four samples: HOPG flakes + benzene (sample A), HOPG flakes + TBAP (sample B), HOPG flakes + Py+Br3− (sample C) and HOPG flakes + BMI+BF4− (sample D) (Fig. S1, Supporting Information). Initially, the comparative stability of each exfoliant was investigated. After sonication in a water bath and standing for 10 h, the EGS in samples A and B were completely precipitated (Fig. 2), which is consistent with previous reports for benzene17,18 which has a π–conjugated structure. TBAP, which has a very bulky structure, also could not exfoliate HOPG because of the steric hindrance of the ammonium cation. However, after standing for 10 h, vials C and D retained their original gray color with no precipitation. The synergistic effect of the π–conjugated planar structures and cationic nature of both Py+Br3− and BMI+BF4− enabled the intercalation of these molecules through the HOPG interlayers and stabilization of the exfoliated graphene sheets.

Illustration of the non-covalent route showing the adsorption of Py+ on the graphene layers, which leads to the formation of an exfoliated graphene dispersion.

(a) Comparative stability of the EGS in (A) benzene, (B) TBAP, (C) Py+ and (D) BMI+. (b), (c), (d) and (e) Typical TEM images of the EGS in samples C and D.
When HOPG and Py+Br3− are mixed with water:ethanol (1:1)and sonicated in a bath-type sonicator, the amphiphilic Py+ attempts to minimize hydrophobic interactions with water by adsorbing on the graphene surfaces via π–π interactions causing the formation of gaps at the edges of the layers, as shown in Fig. 1. In effect, the Py+ molecules act as “molecular wedges” that move deeper and deeper into the interlayers of the graphite with continuing sonication, ultimately leading to complete separation of the layers into sheets. The electrostatic repulsion between the Py+ molecules also reduces the agglomeration of the graphene sheets and assists in maintaining a stable suspension. In fact, the graphene dispersion in water medium was found to be stable for more than a year without any significant precipitation.
Next, to characterize the structure of the EGS, samples C and D were analyzed using transmission electron microscopy (TEM). Low-resolution TEM images are shown in Fig. 2b and 2d, respectively. Large quantities of sub-micrometer-scale EGS can be seen over the entire TEM grid and depending on the level of transparency, wrinkles can be observed on the EGS surfaces. The high-resolution TEM (HR-TEM) images shown in Fig. 2c and 2e reveal that single-layered EGS were produced using Py+ and BMI+, respectively. The diffraction pattern was shown in Fig. S2. In Sample C, 0.85 nm-thick graphene sheets ranging in size from several sub-micrometers to micrometers were finely distributed over a Si wafer support, as determined by atomic force microscopy (Fig. S3), indicating that the HOPG was exfoliated to form single-layered graphene sheets. The average flake size of EGS was 174 ± 105 nm (Fig. S4). Such a thickness is in good agreement with the formation of graphene (≤1 nm)17,19,20. The yield of single-layered and multi-layered graphene was approximately 75% and 25%, respectively, as shown in Table S1. This demonstrates that high-percentage single-layered graphene can be prepared more readily than published results claim21,22,23. Notably, the diluted graphene dispersion enabled the microscopy studies because extensive agglomeration was avoided.
The stability of the EGS suspensions was further investigated by evaluating the transmittance of the dispersed graphene sheets as a function of time. In Fig. 3a, it can be seen that the graphite sheets in sample C remained well-dispersed after standing for 10 h, while the graphite in sample B precipitated within 10 h. Moreover, no increase in the transmittance of samples C and D was observed, even after standing for ten days (Fig. 3b). These results are in good agreement with the images shown in Fig. 2a.

(a) Transmittance variation of EGS after sonicating for 0 (solid line) and 24 h (dot line). Red line: sample B; Black line: sample C. (b) Stability of samples A–D; dependence of the transmittance at 550 nm. Symbols: sample A ( ); sample B (
); sample B ( ); sample C (
); sample C ( ); and sample D (
); and sample D ( ). (c) UV-Vis spectra of aqueous solutions of sample C (λmax = 272 nm) and sample D (λmax = 262 nm).
). (c) UV-Vis spectra of aqueous solutions of sample C (λmax = 272 nm) and sample D (λmax = 262 nm).
Ultraviolet-visible (UV-vis) spectra were obtained to further characterize the electronic conjugation of the EGS samples, which are shown in Fig. 3c that samples C and D showed absorption maxima at 272 and 262 nm, respectively. This suggests that the graphene structure was not converted to graphene oxide24 and the π–conjugated network within the EGS remained the same as that of the pristine graphene suspension. Moreover, the peak in the spectrum for sample C was red-shifted by 10 nm compared to that for sample D, suggesting that the conjugated structure of the EGS in sample C is superior to that of the EGS in sample D. This result also reveals that the level of electronic conjugation in the EGS can be manipulated by selecting an appropriate solvent, thus providing a method for tailoring the functionality of EGS. The synergistic effect of the π–conjugated planar structures and cationic nature of sample C and sample D enabled the intercalation of these molecules through the HOPG interlayer and stabilization of the EGSs. However, sample D had a high boiling point and high viscosity, making it hard to for the graphene to be deposited on surfaces which limits its further electronic application. Next, the statistical distribution of the diameter of the EGS in samples C and D was determined to be ~822 and ~332 nm, respectively, using dynamic light scattering (DLS) (Fig. S5), which is considered to be the most-probable hydrodynamic diameter of an equivalent sphere characterized by the rolling of the EGS.
To understand the chemical composition of the EGS, we employed high-resolution X-ray photoelectron spectroscopy (XPS). The high-resolution core-level C 1s XPS spectrum of the HOPG used in this study and sample C were obtained. The spectrum for HOPG had a narrow peak distribution at 284.7 eV, corresponding to the sp2-hybridized carbon bonding of the aromatic ring (Fig. 4a), while in the spectrum of sample C (Fig. 4b) after Shirley background subtraction, the XPS peaks were fitted with Gaussian and Lorentzian character of 20% and 80%, respectively. The sp2-hybridized carbon bonding of an aromatic ring and a defective C 1s were centered at 284.7 and 285.0 eV, respectively and had a full width half-maximum (fwhm) of 1.4 eV25,26. The ratio of I285.0/I284.7 is a reliable indicator of changes in the sp2 chemical bonding in graphene due to the formation of small flakes after sonication.The I285.0/I284.7 values for HOPG and sample C were 0.18 and 0.30, respectively, indicating that the HOPG was exfoliated to many graphene sheets. Furthermore, in Fig. 4b, the fitted peak at 285.9 eV indicates the presence of C = N bonds, supporting the adsorption of Py+ on the surface of the EGS. The presence of the Py+ moiety can also be identified in the N 1s high-resolution XPS spectrum (Fig. S6). The N 1s peak for the EGS may be due to the presence of the physisorbed Py+ on the surfaces of the EGS27. Finally, the fitted peak appearing at the highest binding energy region was typically much broader (fwhm ~ 2 eV) than the peaks of the components involved in sp2-hybridized carbon bonding. The component at 287.0 eV could be attributed either to C–N configurations28,29,30,31 or C–O structural defects in the graphene sheets. To clearly verify these two possibilities, a Raman spectroscopic analysis was performed.

Characterization of EGS.
XPS spectra of C 1s for (a) pristine HOPG. (b) sample C (EGS + Py+). Raman spectra for (c) pristine HOPG and (d) sample C. (Inset) 2D peak spectrum. Note that for the spectra of the pristine HOPG and sample C, the D band is absent, revealing that virtually no basal plane defects exist. The samples were prepared by dropcasting on a SiO2 wafer followed by drying of the solvent under ambient conditions at room temperature.
In the Raman spectrum of graphene, the peak at approximately 1300 cm−1 is the so-called disorder-induced mode (D band) and the intensity of this band indicates the abundance of defects on the graphene surface. The characteristic C–C stretching in graphene is a Raman-allowed tangential mode (G band) and is an intrinsic characteristic of sp2 carbons. In the Raman spectrum of HOPG, an intense G band peak centered at 1580 cm−1 is observed, but no D band at ~1350 cm−1 is detected (Fig. 4c), while in the spectrum of sample C, an intense peak at ~1575 cm−1 (G band) and ~2675 cm−1 (2D band) can be seen (Fig. 4d), indicating that the HOPG was exfoliated to graphene sheets32,33. Surprisingly, no D band was observed in the Raman spectrum of sample C. The intensity ratio ID/IG is a reliable indicator of the conversion of sp2 carbons to defect forms of carbon. The value of ID/IG for both the HOPG and sample C was 0.004, indicating that no C–O bonds or basal plane defects were present on the surface of the in-plane sp2 carbons. These results confirm that no defects were introduced on the sample C graphene sheets exfoliated in water. The D band (~1350 cm−1) intensity that was detected for the EGSs was quite different from that of the stabilizer-assisted exfoliated graphene with the significant D peak structure defects21,22,23, indicating that the EGSs still preserved their graphene structure. Therefore, the results of the Raman analysis clearly demonstrate that the XPS peak for sample C at 287.0 eV corresponds to C–N bonds rather than C–O configurations. Moreover, Fourier transform infrared-attenuated total reflectance (FTIR-ATR) spectroscopy and thermal gravimetric analysis (TGA) measurements showed that there were no detectable functional groups in the as-prepared EGS (Fig. S7 and S8, respectively). This further confirmed that the fabrication process did not result in the generation of C–O bonds on the EGS.
To further explore the electrical performance of the defect-free and non-oxidized EGS, a uniform EGS paper was readily prepared on a membrane via vacuum-filtration of the sample C suspension. Fig. 5 shows that an electrical conductivity of ~600 S/cm was obtained. The EGS paper was annealed using an electric current flow34. After annealing at a constant power of 0.8 W for 10 s, the electrical conductivity of the EGS paper exhibited an order of magnitude enhancement. This enhancement of the conductivity indicates that the Py+ remaining in the graphene paper were removed during the annealing process. Moreover, excellent sample-to-sample (<5%) reproducibility for planar contact was obtained for different graphene papers due to the increased homogeneity of their surfaces (Fig. 5). The annealing time dependence of the EGS paper conductivity is shown in Fig. S9. After gradually ramping-up, the conductivity of the EGS paper reached saturation as a result of the geometric structure being reorganized between each EGS35. However, when the annealing time was increased to a certain degree, the conductivity of the EGS paper dropped significantly. This may be explained by the probable deterioration of the EGS paper occurring at an ambient condition. Notably, a conductivity of 5130 S/cm was obtained for the annealed EGS paper. To the best of our knowledge, this value is the highest reported value for conductive graphene films prepared from EGS. In fact, the electrical conductivity of the prepared EGS paper was two to five times greater than previously reported results12,36, only two times less than that of in-plane graphite (104 S/cm)37 and nearly 20 times greater than that of a single graphene sheet (60 S/cm)38, which is a prominent 2D material. This result suggests that the Py+-assisted EGS retains the properties of pristine graphene. The superior electrical conductivity may be due to effective π−π stacking between the EGS, which may be beneficial for promoting 3D conductive pathways between each graphene layer.

Electrical characterization of the conducting graphene paper.
(1) with annealing treatment and (2) without annealing treatment.
Finally, the EGS were used as a hole transport layer (HTL) and their performance was compared to that of the commercial HTL material N,N-di(naphthalene-1-yl)-N,N-diphenylbenzidine (NPB), in a basic organic light-emitting diode (OLED) using tris-8-hydroxyquinolinealuminum (Alq3) as the emissive layer (Fig. 6a). For convenience, the OLEDs with graphene sheets as the HTLs and the OLEDs with NPB as the HTLs are referred to as graphene-LED and NPB-LED, respectively. The current density-voltage and luminance-voltage characteristics are shown in Fig. 6b and the luminance efficiency-voltage characteristics are shown in Fig. 5c. As can be seen in Fig. 6b, the phenomenon of abnormal negative differential resistance (NDR), which is often observed in organic devices and typically attributed to ferroelectricity or bias-induced carbon filaments in organic materials, occurred in both devices39,40. With the stacked graphene sheets as the HTL the graphene-LED showed a higher leakage current, probably due to the film discontinuity. However, a lower light-turn-on voltage of 4.9 V is clear evidence that the exfoliated graphene sheets are more beneficial for hole injection than NPB. The luminance-voltage properties of the graphene-LED device were one to two orders of magnitude greater than those of the NPB-LED device. This enhancement implies that the luminance is governed by the doping of the non-oxidized EGS. Considering the reported work function (4.5 eV) of graphene, it is difficult to understand the mechanism for the smaller energy barrier at the graphene/Alq3 interface. In a previous study, by adding an ionic liquid to a ZnO electron transport layer, modified polymer LEDs with significantly improved performance were obtained41. In our case, we assume that the piling up of Py+ at the graphene/Alq3 interface may act as an interface dipole layer with the positive pole pointing toward the graphene and the negative pole pointing toward Alq342. Hence, both the highest occupied molecular orbital of Alq3 and the work function of graphene would be affected and result in the reduction of the hole injection barrier. The maximum luminance of the graphene-LED was 3515 cd/m2 around 7.0 V. In Fig. 6c the luminance efficiency in the graphene-LED was approximately two times of magnitude greater than that in the NPB-LED during device operation. We anticipate that with all the optimal parameters to the device, the high performance of graphene-LED should be possible.

(a) Schematic of the hole-transport process in an OLED from an EGS hole transport layer as an alternative to a conventional NPB HTL. (b) Current density-voltage, luminance-voltage and (c) luminance efficiency-voltage characteristics of an NPB-LED and graphene-LED. (red: graphene-LED; black: NPB-LED).
Discussion
In conclusion, we developed and demonstrated a universal route for the preparation of single-layered graphene sheets in water by choosing exfoliants with a π−conjugated cationic structure. The EGS produced using Py+ as the exfoliant retained the properties of pristine graphene. Defect-free and non-oxidized graphene exhibited an ultra-high conductivity value of ~5100 S/cm, which is only two times inferior to that of in-plane graphite (104 S/cm). Moreover, the EGS can be prepared from HOPG under ambient conditions without the need for rigorous redox chemical reactions. Furthermore, the suspended graphene sheets can be stabilized in water for a year without significant agglomeration. The HTL prepared from the EGS exhibited an enhanced luminance of one to two orders of magnitude compared to that of NPB in OLEDs at low voltage. Thus, it is possible to enhance device luminance at a low operating voltage. These results may be expected for the high-throughput and low-cost production of high-conductivity EGS, thus making the practical application of graphene-based electronics and graphene-composites possible.
Methods
Materials
HOPG (highly oriented pyrolytic graphite, ZYH grade) was purchased from SPI supplies. Benzene (Merck), 1-butyl-3-methylimidazolium tetrafluoroborate (Sigma-Aldrich), pyridinium tribromide (TCI) and TBAP were used to prepare the EGS. Acetone and ethanol were purchased from Sigma-Aldrich and used without further purification.
Preparation of the graphene suspension
Graphene dispersions were prepared by sonicating 2 mg HOPG in 10 ml water:ethanol (1:1) with 1 M Py+ for 45 min using a Branson 3510R-DTH bath ultrasonicator. The prepared EGS concentration in water was 0.04 mg/mL. The graphene suspension was used for spectroscopic analysis and stability tests.
Preparation of the graphene paper
To fabricate the graphene paper, the solution of EGS was filtered through a filter paper or a polyvinylidene fluoride membrane filter (47 mm in diameter, 0.22 μm pore size; Millipore) and then the water was allowed to evaporate by air drying.
Characterization of the exfoliated graphene and graphene paper
Measurement of the absorbance spectra, the pristine state of the dispersed graphene and the chemical composition were obtained via UV-vis spectroscopy (UV 300, UNICAM) and XPS (PHI 1600, Physical Electronics), respectively. The morphology and structure of EGS were investigated using TEM (JEOL JEM-2100). A tapping mode AFM (Veeco Metrology Group/Digital Instruments, Santa Barbara, CA) was used to characterize the thickness of the EGS. Raman spectra were recorded from 2000 to 1000 cm−1 using a high-resolution dispersive Raman microscope (Thermo DXR; excitation at 532 nm). A ZetaSizer (Nano-90S, Malvern) was employed for characterization of the size distribution of the EGS and the diameters of the nanosheets were calculated using the built-in instrument software (Zetasizer DTS). FTIR-ATR measurements were recorded using a Perkin Elmer Frontier. The stability analyses were conducted using a TGA instrument (Q500, TA instrument). The four-point probe method was used to measure the electrical conductivity of the graphene papers. The probe-to-probe spacing was 1.6 mm. The thickness of each filtered EGS films was characterized by the SEM. A Keithley 2400 source meter was used to provide a DC voltage from −0.02 to 0.02 V.
Device fabrication
The HTLs, graphene and NPB, were spin-coated at 3000 rpm and thermally-coated on patterned ITO/glass substrates, respectively. The spin-coating of graphene was repeated two times to obtain the optimal thickness. Subsequently, the Alq3 emissive layer, LiF and Al cathode were thermally-coated on the HTLs to complete the device structure. All of the devices were characterized and preserved in a nitrogen-filled glove box with oxygen and water vapor contents less than 1 ppm. The current density-voltage and the luminance-voltage curves were measured using a combination of a silicon photodetector, a Keithley 2400 source meter and a Keithley 2000 multimeter. The calibration of the absolute luminance was carried out using a PR-650 SpectraScan colorimeter.
References
Novoselov, K. S. et al. A Roadmap for Graphene. Nature 490, 192–200 (2012).
Wassei, J. K. & Kaner, R. B. Oh, the Places You'll Go with Graphene. Acc Chem Res DOI: 10.1021/ar300184v, (2013).
Su, C. & Loh, K. P. Carbocatalysts: Graphene Oxide and Its Derivatives. Acc Chem Res DOI: 10.1021/ar300118v, (2013).
James, D. K. & Tour, J. M. Graphene: Powder, Flakes, Ribbons and Sheets. Acc Chem Res DOI: 10.1021/ar300127r, (2013).
Georgakilas, V. et al. Functionalization of Graphene: Covalent and Non-Covalent Approaches, Derivatives and Applications. Chem Rev 112, 6156–6214 (2012).
Li, X. et al. Large-Area Synthesis of High-Quality and Uniform Graphene Films on Copper Foils. Science 324, 1312–1314 (2009).
Sutter, P. W., Flege, J.-I. & Sutter, E. A. Epitaxial Graphene on Ruthenium. Nature Mater 7, 406–411 (2008).
Berger, C. et al. Electronic Confinement and Coherence in Patterned Epitaxial Graphene. Science 312, 1191–1196 (2006).
Chen, C. et al. Performance of Monolayer Graphene Nanomechanical Resonators with Electrical Readout. Nature Nanotech 4, 861–867 (2009).
Bunch, J. S. et al. Electromechanical Resonators from Graphene Sheets. Science 315, 490–493 (2007).
Novoselov, K. S. et al. Electric Field Effect in Atomically Thin Carbon Films. Science 306, 666–669 (2004).
Park, K. H. et al. Exfoliation of Non-Oxidized Graphene Flakes for Scalable Conductive Film. Nano Lett 12, 2871–2876 (2012).
Edwards, R. S. & Coleman, K. S. Graphene Synthesis: Relationship to Applications. Nanoscale 5, 38–51 (2013).
Coleman, J. N. Liquid Exfoliation of Defect-Free Graphene. Acc Chem Res 46, 14–22 (2013).
Nardecchia, S. et al. Three Dimensional Macroporous Architectures and Aerogels Built of Carbon Nanotubes and/or Graphene: Synthesis and Applications. Chem Soc Rev 42, 794–830 (2013).
Hernandez, Y. et al. High-Yield Production of Graphene by Liquid-Phase Exfoliation of Graphite. Nature Nanotech 3, 563–568 (2008).
Hamilton, C. E. et al. High-Yield Organic Dispersions of Unfunctionalized Graphene. Nano Lett 9, 3460–3462 (2009).
Bourlinos, A. B. et al. Liquid-Phase Exfoliation of Graphite Towards Solubilized Graphenes. Small 5, 1841–1845 (2009).
Nemes-Incze, P., Osvátha, Z., Kamarásb, K. & Biró, L. P. Anomalies in thickness measurements of graphene and few layer graphite crystals by tapping mode atomic force microscopy. Carbon 46, 1435–1442 (2008).
Graf, D. et al. Spatially Resolved Raman Spectroscopy of Single- and Few-Layer Graphene. Nano Lett 7, 238–242 (2007).
Lotya, M. et al. Liquid Phase Production of Graphene by Exfoliation of Graphite in Surfactant/Water Solutions. J Am Chem Soc 131, 3611–3620 (2009).
Lotya, M. et al. High-Concentration, Surfactant-Stabilized Graphene Dispersions. ACS Nano 4, 3155–3162 (2010).
Parviz, D. et al. Dispersions of Non-Covalently Functionalized Graphene with Minimal Stabilizer. ACS Nano 6, 8857–8867 (2012).
Li, D. et al. Processable Aqueous Dispersions of Graphene Nanosheets. Nature Nanotech 3, 101–105 (2007).
An, X. et al. Stable Aqueous Dispersions of Noncovalently Functionalized Graphene from Graphite and their Multifunctional High-Performance Applications. Nano Lett 10, 4295–4301 (2010).
Estrade-Szwarckopf, H. XPS Photoemission in Carbonaceous Materials: A “Defect” Peak beside the Graphitic Asymmetric Peak. Carbon 42, 1713–1721 (2004).
Wang, X. et al. Direct Exfoliation of Natural Graphite into Micrometre Size Few Layers Graphene Sheets using Ionic Liquids. Chem Commun 46, 4487–4489 (2010).
Sampath, S. et al. Direct Exfoliation of Graphite to Graphene in Aqueous Media with Diazaperopyrenium Dications. Adv Mater 25, 2740–2745 (2013).
Zhou, X. et al. Dispersion of Graphene Sheets in Ionic Liquid [bmim][PF6] Stabilized by an Ionic Liquid Polymer. Chem Commun 46, 386–388 (2010).
Jeong, H.-K. et al. Evidence of Graphitic AB Stacking Order of Graphite Oxides. J Am Chem Soc 130, 1362–1366 (2008).
Stankovich, S. et al. Stable Aqueous Dispersions of Graphitic Nanoplatelets via the Reduction of Exfoliated Graphite Oxide in the presence of Poly(Sodium 4-Styrenesulfonate). J Mater Chem 16, 155–158 (2006).
Nuvoli, D. et al. High Concentration Few-Layer Graphene Sheets Obtained by: iquid Phase Exfoliation of Graphite in Ionic Liquid. J Mater Chem 21, 3428–3431 (2011).
Wu, Y. et al. Efficient and Large-Scale Synthesis of Few-Layered Graphene Using an Arc-Discharge Method and Conductivity Studies of the Resulting Films. Nano Res 3, 661–669 (2010).
Rogozin, A. et al. Annealing of Indium Tin Oxide Films by Electric Current: Properties and Structure Evolution. Appl Phys Lett 89, 061908 (2006).
Paulson, S. et al. Tunable Resistance of a Carbon Nanotube-Graphite Interface. Science 290, 1742–1744 (2000).
Geng, X. et al. Interlayer Catalytic Exfoliation Realizing Scalable Production of Large-Size Pristine Few-Layer Graphene. Sci Rep 3, 1134; 10.1038/srep01134 (2013).
Allen, M. J., Tung, V. C. & Kaner, R. B. Honeycomb Carbon: A Review of Graphene. Chem Rev 110, 132–145 (2010).
Du, X., Skachko, I., Barker, A. & Andrei, E. Y. Approaching Ballistic Transport in Suspended Graphene. Nature Nanotech 3, 491–495 (2008).
Naber, R. C. G. et al. Organic Nonvolatile Memory Devices Based on Ferroelectricity. Adv Mater 22, 933–945 (2010).
Chen, Y.-C. et al. Bistable Resistive Switching Characteristics of Poly(2-hydroxyethyl methacrylate) Thin Film Memory Devices. Appl Phys Exp 4, 054204 (2011).
Brine, H., Sánchez-Royo, J. F. & Bolink, H. J. Ionic Liquid Modified Zinc Oxide Injection Layer for Inverted Organic Light-Emitting Diodes. Org Electron 14, 164–168 (2013).
Crispin, X. et al. Characterization of the Interface Dipole at Organic/Metal Interfaces. J Am Chem Soc 124, 8131–8141 (2002).
Acknowledgements
The authors gratefully acknowledge funding from the NSC, Taiwan (101-2113-M-143-003-MY2) and National Taitung University for financial and research support. We also express our gratitude to Dr. S.-Y. Lin, Dr. C.-S. Yang (Center for Nanomedicine Research, National Health Research Institute, Taiwan) for the help of Raman measurements and Dr. C.-T. Kuo for conducting DLS measurement. Thanks to Ms. S.-J. Ji, C.-Y. Chien and Hsu, S.W. of Precious Instrument Center (NTU) for the assistance in SEM, TEM and TGA experiments.
Author information
Authors and Affiliations
Contributions
I.W.P.C. conceived and designed the exfoliation experiments. S.H.S.J. and Y.W.Z. performed the dispersion experiments. C.Y.H. fabricated OLED devices. I.W.P.C. wrote the paper. All the authors discussed the results and commented on the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Chen, IW., Huang, CY., Jhou, SH. et al. Exfoliation and Performance Properties of Non-Oxidized Graphene in Water. Sci Rep 4, 3928 (2014). https://doi.org/10.1038/srep03928
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep03928
This article is cited by
-
A versatile route to edge-specific modifications to pristine graphene by electrophilic aromatic substitution
Journal of Materials Science (2020)
-
Graphite to Graphene: Green Synthesis Using Opuntia ficus-indica
Journal of Electronic Materials (2019)
-
Surfactant molecules make liquid phase exfoliated graphene a switching element for resistive random access memory applications
Journal of Materials Science: Materials in Electronics (2018)
-
Large-Scale Production of Large-Size Atomically Thin Semiconducting Molybdenum Dichalcogenide Sheets in Water and Its Application for Supercapacitor
Scientific Reports (2016)
-
Nanoparticle-Mediated Physical Exfoliation of Aqueous-Phase Graphene for Fabrication of Three-Dimensionally Structured Hybrid Electrodes
Scientific Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
