
Abstract
Toll-like receptors (TLRs), as innate immunity sensors, play critical roles in immune responses. Six SNPs of TLR3, TLR7 and TLR8 were genotyped to determine their associations with systemic lupus erythematosus (SLE) and clinical manifestations of SLE. TLR7 SNP rs3853839 was independently associated with SLE susceptibility in females (G vs. C: p = 0.0051). TLR7 rs3853839-G (G vs. C: p = 0.0100) and TLR8 rs3764880-G (recessive model: p = 0.0173; additive model: p = 0.0161) were associated with pericardial effusion in females relative to healthy females. Anti-SSA positive cases were more likely to have the dominant TLR7 rs179010-T allele than normal controls (p = 0.0435). TLR3 rs3775296-T was associated with photosensitivity (p = 0.0020) and anemia (p = 0.0082). The “G-G” haplotype of TLR7 rs3853839 and TLR8 rs3764880 increased risk of SLE in females (age adjusted p = 0.0032). These findings suggest that TLR variations that modify gene expression affect risk for SLE susceptibility, clinical phenotype development and production of autoantibodies.
Similar content being viewed by others
Introduction
System lupus erythematosus (SLE) is a systemic autoimmune disease characterized by widespread loss of immune tolerance to self-antigens. Genetic and environmental factors contribute to the development of SLE and patients typically experience alternating periods of flare-up and remission1. The generation of numerous autoantibodies that react with self-nuclear and -cytoplasmic antigens is associated with the dysfunction of multiple organ systems2,3,4,5. The genetic transmission and patterns of inheritance of SLE have not yet been elucidated. In particular, it has been difficult to identify specific genetic polymorphisms associated with SLE due to the presence of polygenic inheritance, extensive genetic heterogeneity, the small size of most genetic studies and the low disease prevalence6,7. Genome-wide association studies (GWASs) of various populations have identified several common immune response pathways and the presence of genetic variants in some ethnic groups that are involved in the pathogenesis of SLE8,9,10,11,12,13. However, genetic dissection of SLE and other autoimmune diseases is difficult because these are complex diseases that involve alterations of multiple biologic pathways14. The advent of modern genomics and the availability of new technologies have made it possible to fine-map candidate genes for SLE and other specific diseases based on knowledge of gene map position and functional relevance15,16,17,18. Such studies may help to identify the roles of candidate genes for SLE and the roles of different genes in the expression of different clinical manifestations4,19,20.
Previous studies have indicated that innate pattern recognition receptors, such as Toll-like receptors (TLRs), play important roles in the development of autoimmunity. TLR proteins are localized on the cell surface or in endosomes and play critical roles in innate immune responses against different pathogens21,22. Internalized nucleic acid immune complexes act as endogenous ligands that activate intracellular TLRs and these initiate several signaling pathways that lead to increased production of type I interferons (IFNs) in plasmacytoid dendritic cells (pDCs)22,23,24,25,26. Increased production of type I IFNs increases apoptosis, neutrophil cell death via neutrophil extracellular trap (NETosis), innate immune signaling and viral infection-induced autoimmunity27,28,29. Aberrant stimulation of the innate immune system through intracellular TLRs may lead to hyperactive immune responses and contribute to the pathogenesis of SLE30,31,32.
TLR3, TLR7 and TLR8 are primarily associated with endosomal membranes and they recognize microbial nucleic acids. TLR3 binds to viral double-stranded RNA (dsRNA) and induces antiviral immune responses by promoting the production of type I IFN and pro-inflammatory cytokines. TLR7 binds to single-stranded RNA (ssRNA) from RNA viruses and triggers pDCs to produce type I IFN. TLR8 is phylogenetically related to TLR7 and also mediates recognition of viral ssRNA. Thus, these 3 TLRs are responsible for pathogen clearance, antigen recognition and induction of cytokine production30,33,34.
Genetic and hormonal factors appear to partially explain the female predominance of SLE35. In particular, TLR7 and TLR8 are on the X chromosome (Xp22.2) and play critical roles in innate immunity and inflammatory responses30. Studies of animal models indicated that TLR7 gene dosage significantly affected the hyperactivity of B cells36,37,38,39,40,41. Mice with a Y-linked autoimmune accelerator (Yaa) that carries an extra copy of TLR7 develop autoimmunity to RNA-associated autoantigens, but mice with a low TLR7 copy number require additional susceptibility loci to develop autoimmunity39,40. In humans, males with Klinefelter syndrome carry an extra X chromosome (47, XXY) and are more likely to develop SLE; females with Turner syndrome lack one X chromosome (45, X) and are less likely to develop SLE42,43. In addition, a study of a Mexican population indicated that increased TLR7 copy number correlated with TLR7 mRNA levels and susceptibility to childhood-onset SLE44 although TLR7 copy number variations (CNVs) are infrequent in human SLE45. Other studies indicated that increased expression of TLRs in peripheral blood mononuclear cells and lymphocytes led increased IFN-α expression in SLE patients46,47,48.
Previous research indicated that a functional TLR7 SNP (rs3853839-G > C) affects TLR7 expression by modulation of microRNA-3148 (miR-3148)49 and that two other intron SNPs (rs5935436 and rs179010) were associated with increased SLE susceptibility45,49. The TLR8 SNP rs3764880 is a functional polymorphism that affects TLR8 transcription and translations of TLR8 isoforms, which leads to the activation difference of NF-κB, is also associated with SLE susceptibility45,49,50,51,52. A study of cell cultures indicated that TLR3 rs3775296 (in the promoter) and rs3775291 (in exon 4) affected TLR3 cell surface expression and localization and subsequently influenced NF-κB cascade induction although the effect of these mutations has not been assessed in clinical studies53. The present study examined the associations of the above mentioned 6 SNPs (3 in TLR7, 1 in TLR8, 2 in TLR3) with SLE and with specific clinical manifestations of SLE.
Results
We investigated the role of 6 SNPs in the susceptibility to SLE (rs3775296 and rs3775291 from TLR3; rs5935436, rs179010 and rs3853839 from TLR7; and rs3764880 from TLR8) by examination of 795 SLE patients (68 males and 727 females) and 1162 healthy controls (513 males and 649 females). The average age of cases was 30.71 years (SD = 11.62, range: 8 to 77 years), 8.55% were males (31.46 ± 12.52 years-old) and 91.45% were females (30.64 ± 11.56 years-old). The healthy controls had a mean age of 40.24 years (SD = 10.88, range 18–64 years), with similar average ages of males and females (40.26 ± 9.26 years vs. 40.23 ± 12.02 years). The age difference of SLE patients and healthy controls was statistically significant for both males and females, so we adjusted for age in the subsequent association analysis. Table 1 shows the clinical characteristics of the 795 SLE patients, with independent statistical analyses for males and females. In the female cases and controls, there were no deviations from HWE in the six candidate SNPs.
Table 2 shows the single-locus associations between the six candidate SNPs and susceptibility to SLE. In males, there were no significant case-control associations after the false discovery rate (FDR) correction, possibly due to the small sample size. In females, there was a significant allelic association between SLE and rs3853839 in TLR7 (PFDR = 0.013, OR = 1.38, 95% CI = 1.12–1.69). Moreover, the risk allele G had a recessive effect in females (GG vs. GC + CC: PFDR = 0.017, OR = 1.44, 95% CI = 1.14–1.83), suggesting that rs3853839 plays a role in development of SLE. We also evaluated the independent contributions of the 6 candidate SNPs (adjusted for age) to SLE risk in females and performed a multivariate logistic regression analysis that included the significant SNPs with the same genetic models. The rs3853839 SNP was the only significant susceptibility marker among the 6 examined SNPs (G vs. C: p = 0.0051, OR = 1.42, 95% CI = 1.11–1.82).
TLR SNP polymorphisms affected SLE phenotype and production of autoantibodies
Patients with SLE are present with heterogeneous clinical features and have significant variations in the severity, nature and spectrum of clinical involvement. Thus, we examined the effect of TLR SNP polymorphisms on SLE clinical parameters and phenotypes. Based on the clinical characteristics of males and females (Table 1), we initially performed two comparisons: (i) allele frequencies of SLE patients with each characteristic (“+” in Tables S1–S3) and SLE patients without the characteristic (“−” in Tables S1–S3); and (ii) allele frequencies of SLE patients with each characteristic and the normal controls (“normal” in Tables S1–S3). In males, there were no significant associations of the 6 candidate SNPs with SLE clinical manifestations after the FDR correction (Table 3, with more details in Tables S1 and S3). Again, this may result from the small sample size.
In females, TLR7 rs3853839 G risk allele was associated with several clinical manifestations of SLE (Table 4). The comparison of phenotype-positive cases and normal controls indicated significant associations of this allele with oral ulcer, arthritis, malar rash, photosensitivity, pericardial effusion, depressed complement, anti-dsDNA, anti-Sm and anti-SSA (PFDR < 0.05 for all comparisons). Multivariate logistic regression analysis of the independent association of each clinical characteristic with rs3853839 indicated that only pericardial effusion was significantly associated with the TLR7 rs3853839 G risk allele. In particular, cases with pericardial effusion were more likely to have the G risk allele than normal controls (p = 0.0100, OR = 2.82, 95% CI = 1.28–6.19). Table S2 shows that anti-SSA was associated with the rs179010 T risk allele (T vs. C: PFDR = 0.0366, OR = 1.47, 95% CI = 1.11–1.94; CT + TT vs. CC: PFDR = 0.0037, OR = 1.93, 95% CI = 1.33–2.81). Multivariate logistic regression analysis also showed that cases who were anti-SSA positive were more likely to carry the dominant rs179010 T risk allele than normal controls (p = 0.0435, OR = 1.33, 95% CI = 1.01–1.75), but cases who were anti-SSA negative had the opposite tendency (p = 0.0299, OR = 0.69, 95% CI = 0.49–0.96).
Although rs3764880 in TLR 8 was not associated with SLE susceptibility, the risk allele G was associated with oral ulcer with significant additive effects (PFDR = 0.0232: OR = 1.66, 95% CI = 1.18–2.35, G vs. A: PFDR = 0.0300, OR = 1.65, 95% CI = 1.17–2.32). However, incorporation of clinical variables with nominal p values below 0.05 into the multivariate logistic regression model indicated that pericardial effusion was the only clinical characteristic associated with rs3764880. In particular, pericardial effusion-positive cases were more likely to carry the rs3764880 G risk allele than normal controls, either with a recessive effect (p = 0.0173, OR = 2.91, 95% CI = 1.21–7.02) or with an additive effect (p = 0.0161, OR = 2.96, 95% CI = 1.22–7.14).
Analysis of the association of TLR3 SNPs with SLE clinical characteristics indicated no significant associations for males (Tables 3 and S3). In females, comparison of phenotype-positive cases with phenotype-negative cases and of phenotype-positive cases with normal controls indicated positive associations of rs3775296 with anemia (Table 4, TT vs. GG + GT: PFDR = 0.0244, OR = 2.41, 95% CI = 1.33–4.39; TT vs. GG + GT: PFDR = 0.0373, OR = 2.11, 95% CI = 1.23–3.65, respectively). Multivariate logistic regression analysis indicated that photosensitivity-negative cases and anemia-negative cases were associated with recessive rs3775296 T risk allele relative to normal controls (photosensitivity: p = 0.0020, OR = 2.40, 95% CI = 1.38–4.19; anemia: p = 0.0082, OR = 0.44, 95% CI = 0.24–0.81).
TLR haplotypes were associated with SLE susceptibility in females
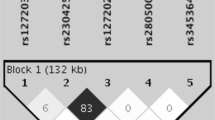
In Figure 1, pair-wise LD measures r2 and D′ for the four TLR7 and TLR8 SNPs rs3853839, rs5935436, rs179010 and rs3764880 in the female healthy controls were presented. We estimated the frequencies of haplotypes formed by these 4 SNPs in females to determine the associations of different haplotypes with susceptibility to SLE. Table 5 shows the 4 inferred haplotypes that had estimated frequencies more than 5%. Performance of haplotype-trait association tests in female SLE patients and healthy controls indicated that the “C-C-C-G” haplotype protected against development of SLE (5.78% in cases vs. 8.55% in controls; permutation p-value = 0.0033, OR = 0.60, 95% CI = 0.44–0.82 after age adjustment). In Table 6 shows the estimates for haplotypes from rs3853839 and rs3764880 SNPs only. These results show that the effect of “C-G” on SLE susceptibility remained significant when we controlled for age (7.08% in cases vs. 8.39% in controls; permutation p-value = 0.0073, OR = 0.62, 95% CI = 0.46–0.83). Nevertheless, females with “G-G” haplotypes of rs3853839 and rs3764880 were significantly more likely to develop SLE (age adjusted p-value = 0.0032, OR = 1.32, 95% CI = 1.10–1.60).

Pair-wise linkage disequilibrium patterns with r2 (left side) and D′ (right side) measures of the four SNPs in TLR7 and TLR8 at Xp22.3 for 649 healthy female controls.
Discussion
Previous research with large samples indicated that the functional TLR7 SNP rs3853839-G > C was significantly associated with SLE in East Asians, especially in males45. The current study identified the rs3853839-G allele of TLR7 as the main susceptibility marker in female SLE patients from Taiwan based on single-locus and multivariate logistic regression analyses. This confirms the role of this SNP in the pathogenesis of SLE. The genetic effect of TLR7 on SLE appears to vary among different ethnic groups45,54,55,56. In particular, a previous study indicated that the TLR7 intronic SNPs rs179019 and rs179010 were associated with SLE, independent of the 3′ UTR SNP rs3853839 in Japanese females54. Other research indicated that the TLR7 SNP rs179008 was not associated with SLE in a European population, but was significantly associated with SLE in Brazilians55,56. Recently, Deng et al. conducted a large trans-ancestral fine-mapping of European Americans, African Americans and Amerindian/Hispanics and identified rs3853839-G as the only genetic risk variant for SLE in the TLR7-TLR8 region, although they did not confirm the male specific association. Notably, rs3853839-G appears to increase risk for SLE in different populations, although there are different frequencies in different ethnic groups. This highlights the critical role of elevated TLR7 expression in the pathogenesis of SLE, which in this case is mediated by slower mRNA degradation due to miR-3148 expression49.
TLR7 and TLR8 contribute to antigen recognition and antibody production in the pathogenesis of SLE. TLR ligands, which are present in viruses and virus-like particles (VLPs), can directly stimulate B cells and B cell responses are integrated with dual antigen-specific B cell receptor (BCR) and TLR engagement57,58. B cells with up-regulated MYD88 expression become more responsive to TLR ligands and promote class-switch recombination59. The inactivation or overexpression of genes that encode TLRs or of molecules that alter TLR signaling provides a bridge between the innate and adaptive immune systems and this is critical to the presence of B cells defects in the pathogenesis of SLE60,61. Moreover, the recognition of endogenous RNA-containing antigens by TLR7/3 may trigger autoreactive B cells in the germinal center and this is accompanied by the suppression of T regulatory cells, leading to disruption of self-tolerance62,63,64. In this regard, numerous innate and adaptive immunity related genes involving IFN-alpha mediated signature pathway as well as T and B cells activation signaling pathways participate in the SLE pathogenesis65. The present study found that TLR7 rs3853839 G risk allele was associated with several clinical manifestations of SLE, including oral ulcer, arthritis, malar rash, photosensitivity, pericardial effusion, depressed complement and anti-dsDNA, anti-Sm and anti-SSA autoantibodies. The TLR7 rs179010 T risk allele was also associated with anti-SSA autoantibodies. These findings suggest that TLR7 may play a key role in autoantibody production because it increases B-cell sensitivity to RNA-containing autoantigens in the development of systemic autoimmunity. However, given the limited sample sizes of our stratified phenotype groups, this finding requires replication by larger future studies.
We also observed that the non-synonymous TLR8 SNP rs3764880-G allele was a risk factor for oral ulcer and pericardial effusion, with significant additive effects. Interestingly, previous research indicated that the TLR8 rs3764880-G allele protected against tissue damage in active tuberculosis and predicted a slower disease course in patients with HIV infections52,66. The TLR8 SNP rs3764880 alters the ATG start codon of TLR8 isoform B into a GTG. A methionine at position 4 of isoform B is used as the start codon for the TLR8-rs3764880G allele, resulting in a truncated TLR8 isoform B with a shorter signal peptide (1038 residues for the TLR8-rs3764880G allele vs. 1041 residues for the TLR8-rs3764880A allele). Immune cells carrying the TLR8-rs3764880G allele had augmented TNFα-responses, but decreased translation of truncated TLR8 isoform B and NF-κB production relative to those carrying the TLR8-rs3764880A allele50,66. Therefore, TLR8 appears to have important roles in autoimmune diseases and in response to infections.
Previous research indicated that TLR7 and TLR8 have closely related functions in immune responses. We observed that the TLR7 and TLR8 SNP haplotype rs5935436-C/rs179010-C/rs3853839-C/rs3764880-G protected against development of SLE, but the effect of rs3853839-C/rs3764880-G on SLE susceptibility remained significant when controlled for age. On the other hand, females with G-G haplotypes of rs3853839 and rs3764880 were significantly more likely to develop SLE. These results indicate that TLR8 may play a regulatory role in TLR7 function in innate immunity, similar to that documented in mice67.
TLR3 recognizes dsRNA and its elevated level in human SLE peripheral blood cells suggests that it may play a key role in IFN signature gene activation22,46,47,48. In addition, previous research indicated that TLR3 aggravated lupus nephritis in lupus-prone mice68. We observed that TLR3 rs3775296-T risk allele had recessive effects on photosensitivity and anemia-negative SLE patients. Thus, our data indicate that TLR3 may play a role in the development of different SLE phenotypes and in antiviral responses that trigger expression of pro-inflammatory genes.
In conclusion, the present study confirmed that certain functional TLR7 and TLR8 variations, particularly TLR7 rs3853839-G > C, modify gene expression and increase the risk for SLE, development of certain SLE phenotypes and production of autoantibodies. These results suggest that TLR7/8 genetic variations are potential biomarkers for prediction of SLE phenotypes and also have implications for the development of therapeutic measures that may prevent various pathological conditions that are characteristic of SLE.
Methods
Characteristics of the study populations
All patients were recruited from the clinics of Chang Gung Memorial Hospital and rheumatology specialists confirmed that all patients fulfilled the 1982 and 1997 American College of Rheumatology (ACR) diagnostic criteria for SLE69. For the purpose of this study, healthy controls were selected following a questionnaire to assure that they did not have any autoimmune disease phenotypes. This study was approved by the ethics committees of Chang Gung Memorial Hospital and all patients provided written informed consent according to the Declaration of Helsinki.
Genomic DNA extraction
Genomic DNA was extracted from EDTA-anticoagulated peripheral blood using the Purgene DNA isolation kit as described previously70.
SNPs genotyping assays
Validated Applied Biosystem TaqMan SNP assays were used for genotype determination of TLRs according the vendor's instructions (Life Technologies, Grand Island, NY). The TaqMan allele discrimination assays were performed on an ABI ViiA 7 Real-time System (Life Technology) and probes were labeled with a fluorescent dye (FAM and VIC).
Statistical analysis
The functional candidate gene approach was used to perform a case-control association study by examination of 795 SLE patients (68 males and 727 females) and 1162 healthy controls (513 males and 649 females). The Hardy-Weinberg equilibrium (HWE) was examined for the 6 selected SNPs using chi-square tests in females with and without SLE. Differences of allele frequencies in each of the 6 SNPs were separately assessed in males and females to investigate the single-locus associations. Additionally, for females, the significance of differences in genotype frequencies were also evaluated and dominant and recessive models were tested for each SNP. The p-values, odds ratios (ORs) and 95% confidence intervals (CIs) were then calculated based on the risk allele identified. TLR 8 and TLR7 are located on the X chromosome (Xp22.2), so female and male data were analyzed separately. Meta-analysis with generation of meta p-values using the Cochran-Mantel-Haenzsel (CMH) method was used to assess the allelic associations of the 6 SNPs in all samples.
We initially stratified clinical phenotypes according to diagnostic criteria to investigate the association of each SNP with SLE clinical manifestations. SLE patients with the phenotype under investigation were classed as “+” cases, those without this phenotype as “−” cases and healthy controls as “normal”. Then, allele frequencies were compared for “+” cases and “−” cases and for “+” cases and “normal” controls. In females, additional analyses also considered additive, dominant and recessive effects for the risk allele of each SNP in order to assess genotype-phenotype associations. To investigate the independent association of SLE clinical characteristics with the six SNPs, multivariate logistic regression analysis was subsequently carried out to identify the independent statistical association of significant phenotypes (identified above) with the six SNPs. The additive, dominant and recessive allele effects for each SNP were modeled as the response variable and the three categories of individuals: “+” cases, “−” cases and “Normal” pertaining to each clinical characteristic were used as the independent variables.
Linkage disequilibrium (LD) patterns of neighboring SNPs on the same chromosome were analyzed by Haploview 4.2 (Broad Institute, Cambridge, MA, USA; http://www.broad.mit.edu/mpg/haploview). Haplotype information was inferred and frequencies were estimated using the HAPLOTYPE procedure in SAS 9.2 (SAS Institute, Cary, NC). Differences in haplotype frequencies were assessed for SLE cases and controls were separately assessed in males and females. The permutation (N = 10,000) p-values of each haplotype were calculated using the expectation-maximization (EM) algorithm, conditional on the other haplotypes, to evaluate the independent association of each category of haplotypes. Benjamini and Hochberg's linear step-up method in the SAS MULTTEST procedure was used to account for multiple testing71. The False Discovery Rate (FDR)-adjusted p-values are defined in a step-up fashion, with less conservative multipliers and control. A corrected p-value (PFDR) less than 0.05 indicated statistical significance.
References
Tsokos, G. C. Systemic lupus erythematosus. N Engl J Med 365, 2110–2121 (2011).
Hughes, T. & Sawalha, A. H. The role of epigenetic variation in the pathogenesis of systemic lupus erythematosus. Arthritis Res Ther 13, 245 (2011).
Pathak, S. & Mohan, C. Cellular and molecular pathogenesis of systemic lupus erythematosus: lessons from animal models. Arthritis Res Ther 13, 241 (2011).
Liu, Z. & Davidson, A. Taming lupus-a new understanding of pathogenesis is leading to clinical advances. Nat Med 18, 871–882 (2012).
Gualtierotti, R., Biggioggero, M., Penatti, A. E. & Meroni, P. L. Updating on the pathogenesis of systemic lupus erythematosus. Autoimmun Rev 10, 3–7 (2010).
Wakeland, E. K., Liu, K., Graham, R. R. & Behrens, T. W. Delineating the genetic basis of systemic lupus erythematosus. Immunity 15, 397–408 (2001).
Lauwerys, B. R. & Wakeland, E. K. Genetics of lupus nephritis. Lupus 14, 2–12 (2005).
Remmers, E. F. et al. STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N Engl J Med 357, 977–986 (2007).
Harley, J. B. et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet 40, 204–210 (2008).
Hom, G. et al. Association of systemic lupus erythematosus with C8orf13-BLK and ITGAM-ITGAX. N Engl J Med 358, 900–909 (2008).
Yang, W. et al. Genome-wide association study in Asian populations identifies variants in ETS1 and WDFY4 associated with systemic lupus erythematosus. PLoS Genet 6, e1000841 (2010).
Han, J. W. et al. Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat Genet 41, 1234–1237 (2009).
Deng, Y. & Tsao, B. P. Genetic susceptibility to systemic lupus erythematosus in the genomic era. Nat Rev Rheumatol 6, 683–692 (2010).
Cui, Y., Sheng, Y. & Zhang, X. Genetic susceptibility to SLE: Recent progress from GWAS. J Autoimmun (2013).
Harley, I. T., Kaufman, K. M., Langefeld, C. D., Harley, J. B. & Kelly, J. A. Genetic susceptibility to SLE: new insights from fine mapping and genome-wide association studies. Nat Rev Genet 10, 285–290 (2009).
Lessard, C. J. et al. Identification of IRF8, TMEM39A and IKZF3-ZPBP2 as susceptibility loci for systemic lupus erythematosus in a large-scale multiracial replication study. Am J Hum Genet 90, 648–660 (2012).
Gateva, V. et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat Genet 41, 1228–1233 (2009).
Yang, W. et al. Meta-analysis followed by replication identifies loci in or near CDKN1B, TET3, CD80, DRAM1 and ARID5B as associated with systemic lupus erythematosus in Asians. Am J Hum Genet 92, 41–51 (2013).
Guerra, S. G., Vyse, T. J. & Cunninghame Graham, D. S. The genetics of lupus: a functional perspective. Arthritis Res Ther 14, 211 (2012).
Patel, D. R. & Richardson, B. C. Dissecting complex epigenetic alterations in human lupus. Arthritis Res Ther 15, 201 (2013).
Akira, S., Uematsu, S. & Takeuchi, O. Pathogen recognition and innate immunity. Cell 124, 783–801 (2006).
Barbalat, R., Ewald, S. E., Mouchess, M. L. & Barton, G. M. Nucleic acid recognition by the innate immune system. Annu Rev Immunol 29, 185–214 (2011).
Ewald, S. E. & Barton, G. M. Nucleic acid sensing Toll-like receptors in autoimmunity. Curr Opin Immunol 23, 3–9 (2011).
Means, T. K. et al. Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J Clin Invest 115, 407–417 (2005).
Bave, U. et al. Fc gamma RIIa is expressed on natural IFN-alpha-producing cells (plasmacytoid dendritic cells) and is required for the IFN-alpha production induced by apoptotic cells combined with lupus IgG. J Immunol 171, 3296–3302 (2003).
Baccala, R., Hoebe, K., Kono, D. H., Beutler, B. & Theofilopoulos, A. N. TLR-dependent and TLR-independent pathways of type I interferon induction in systemic autoimmunity. Nat Med 13, 543–551 (2007).
Ronnblom, L., Alm, G. V. & Eloranta, M. L. The type I interferon system in the development of lupus. Semin Immunol 23, 113–121 (2011).
Garcia-Romo, G. S. et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med 3, 73ra20 (2011).
Elkon, K. B. & Wiedeman, A. Type I IFN system in the development and manifestations of SLE. Curr Opin Rheumatol 24, 499–505 (2012).
Bauer, S., Pigisch, S., Hangel, D., Kaufmann, A. & Hamm, S. Recognition of nucleic acid and nucleic acid analogs by Toll-like receptors 7, 8 and 9. Immunobiology 213, 315–328 (2008).
Kontaki, E. & Boumpas, D. T. Innate immunity in systemic lupus erythematosus: sensing endogenous nucleic acids. J Autoimmun 35, 206–211 (2010).
Christensen, S. R. & Shlomchik, M. J. Regulation of lupus-related autoantibody production and clinical disease by Toll-like receptors. Semin Immunol 19, 11–23 (2007).
Heil, F. et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 303, 1526–1529 (2004).
Gilliet, M., Cao, W. & Liu, Y. J. Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat Rev Immunol 8, 594–606 (2008).
Rubtsov, A. V., Rubtsova, K., Kappler, J. W. & Marrack, P. Genetic and hormonal factors in female-biased autoimmunity. Autoimmun Rev 9, 494–498 (2010).
Pisitkun, P. et al. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science 312, 1669–1672 (2006).
Subramanian, S. et al. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc Natl Acad Sci U S A 103, 9970–9975 (2006).
Santiago-Raber, M. L. et al. Evidence for genes in addition to Tlr7 in the Yaa translocation linked with acceleration of systemic lupus erythematosus. J Immunol 181, 1556–1562 (2008).
Hwang, S. H. et al. B cell TLR7 expression drives anti-RNA autoantibody production and exacerbates disease in systemic lupus erythematosus-prone mice. J Immunol 189, 5786–5796 (2012).
Moisini, I. et al. The Yaa locus and IFN-alpha fine-tune germinal center B cell selection in murine systemic lupus erythematosus. J Immunol 189, 4305–4312 (2012).
Walsh, E. R. et al. Dual signaling by innate and adaptive immune receptors is required for TLR7-induced B-cell-mediated autoimmunity. Proc Natl Acad Sci U S A 109, 16276–16281 (2012).
Scofield, R. H. et al. Klinefelter's syndrome (47,XXY) in male systemic lupus erythematosus patients: support for the notion of a gene-dose effect from the X chromosome. Arthritis Rheum 58, 2511–2517 (2008).
Cooney, C. M. et al. 46,X,del(X)(q13) Turner's syndrome women with systemic lupus erythematosus in a pedigree multiplex for SLE. Genes Immun 10, 478–481 (2009).
Garcia-Ortiz, H. et al. Association of TLR7 copy number variation with susceptibility to childhood-onset systemic lupus erythematosus in Mexican population. Ann Rheum Dis 69, 1861–1865 (2010).
Shen, N. et al. Sex-specific association of X-linked Toll-like receptor 7 (TLR7) with male systemic lupus erythematosus. Proc Natl Acad Sci U S A 107, 15838–15843 (2010).
Komatsuda, A. et al. Up-regulated expression of Toll-like receptors mRNAs in peripheral blood mononuclear cells from patients with systemic lupus erythematosus. Clin Exp Immunol 152, 482–487 (2008).
Wong, C. K. et al. Activation profile of Toll-like receptors of peripheral blood lymphocytes in patients with systemic lupus erythematosus. Clin Exp Immunol 159, 11–22 (2010).
Midgley, A., Thorbinson, C. & Beresford, M. W. Expression of Toll-like receptors and their detection of nuclear self-antigen leading to immune activation in JSLE. Rheumatology (Oxford) 51, 824–832 (2012).
Deng, Y. et al. MicroRNA-3148 modulates allelic expression of toll-like receptor 7 variant associated with systemic lupus erythematosus. PLoS Genet 9, e1003336 (2013).
Gantier, M. P. et al. Genetic modulation of TLR8 response following bacterial phagocytosis. Hum Mutat 31, 1069–1079 (2010).
Cervantes, J. L., Weinerman, B., Basole, C. & Salazar, J. C. TLR8: the forgotten relative revindicated. Cell Mol Immunol 9, 434–438 (2012).
Davila, S. et al. Genetic association and expression studies indicate a role of toll-like receptor 8 in pulmonary tuberculosis. PLoS Genet 4, e1000218 (2008).
Ranjith-Kumar, C. T. et al. Effects of single nucleotide polymorphisms on Toll-like receptor 3 activity and expression in cultured cells. The Journal of biological chemistry 282, 17696–17705 (2007).
Kawasaki, A. et al. TLR7 single-nucleotide polymorphisms in the 3′ untranslated region and intron 2 independently contribute to systemic lupus erythematosus in Japanese women: a case-control association study. Arthritis Res Ther 13, R41 (2011).
Sanchez, E. et al. Investigation of TLR5 and TLR7 as candidate genes for susceptibility to systemic lupus erythematosus. Clin Exp Rheumatol 27, 267–271 (2009).
dos Santos, B. P. et al. TLR7/8/9 polymorphisms and their associations in systemic lupus erythematosus patients from southern Brazil. Lupus 21, 302–309 (2012).
Lau, C. M. et al. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J Exp Med 202, 1171–1177 (2005).
Rawlings, D. J., Schwartz, M. A., Jackson, S. W. & Meyer-Bahlburg, A. Integration of B cell responses through Toll-like receptors and antigen receptors. Nat Rev Immunol 12, 282–294 (2012).
Giltiay, N. V., Chappell, C. P. & Clark, E. A. B-cell selection and the development of autoantibodies. Arthritis Res Ther 14 Suppl 4, S1 (2012).
Avalos, A. M., Busconi, L. & Marshak-Rothstein, A. Regulation of autoreactive B cell responses to endogenous TLR ligands. Autoimmunity 43, 76–83 (2010).
Green, N. M. & Marshak-Rothstein, A. Toll-like receptor driven B cell activation in the induction of systemic autoimmunity. Semin Immunol 23, 106–112 (2011).
Rubtsov, A. V. et al. Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c(+) B-cell population is important for the development of autoimmunity. Blood 118, 1305–1315 (2011).
Green, N. M., Moody, K. S., Debatis, M. & Marshak-Rothstein, A. Activation of autoreactive B cells by endogenous TLR7 and TLR3 RNA ligands. J Biol Chem 287, 39789–39799 (2012).
Hackl, D., Loschko, J., Sparwasser, T., Reindl, W. & Krug, A. B. Activation of dendritic cells via TLR7 reduces Foxp3 expression and suppressive function in induced Tregs. Eur J Immunol 41, 1334–1343 (2011).
Bronson, P. G., Chaivorapol, C., Ortmann, W., Behrens, T. W. & Graham, R. R. The genetics of type I interferon in systemic lupus erythematosus. Curr Opin Immunol 24, 530–537 (2012).
Oh, D. Y. et al. A functional toll-like receptor 8 variant is associated with HIV disease restriction. J Infect Dis 198, 701–709 (2008).
Demaria, O. et al. TLR8 deficiency leads to autoimmunity in mice. J Clin Invest 120, 3651–3662 (2010).
Patole, P. S. et al. Viral double-stranded RNA aggravates lupus nephritis through Toll-like receptor 3 on glomerular mesangial cells and antigen-presenting cells. J Am Soc Nephrol 16, 1326–1338 (2005).
Hochberg, M. C. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 40, 1725 (1997).
Chen, J. Y. et al. Association of a transmembrane polymorphism of Fcgamma receptor IIb (FCGR2B) with systemic lupus erythematosus in Taiwanese patients. Arthritis Rheum 54, 3908–3917 (2006).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society. Series B (Methodological), 289–300 (1995).
Acknowledgements
We greatly appreciate the Shin Chu Blood Donor Center for providing samples. This work was supported by National Science Council, Taiwan and CMRPG 391813 from Chang Gung Memorial Hospital.
Author information
Authors and Affiliations
Contributions
C.M.W. and J.Y.C. performed study design, manuscript preparation and coordination. S.W.C. performed statistical analysis, data interpretation and manuscript preparation. T.C.C. participated in the statistical analysis. Y.J.J., J.C.L., H.H.H. and P.Y. participated in sample acquisition and data interpretation. J.W. conceived of the study, participated in its design and helped to draft the manuscript. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Genetic variations in Toll-like receptors (TLRs 3/7/8) are associated with systemic lupus erythematosus in a Taiwanese population
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Wang, CM., Chang, SW., Wu, YJ. et al. Genetic variations in Toll-like receptors (TLRs 3/7/8) are associated with systemic lupus erythematosus in a Taiwanese population. Sci Rep 4, 3792 (2014). https://doi.org/10.1038/srep03792
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep03792
This article is cited by
-
Association of X-linked TLR-7 gene polymorphism with the risk of knee osteoarthritis: a case–control study
Scientific Reports (2022)
-
TLR7 gain-of-function genetic variation causes human lupus
Nature (2022)
-
Pathogen-associated selection on innate immunity genes (TLR4, TLR7) in a neotropical rodent in landscapes differing in anthropogenic disturbance
Heredity (2020)
-
COVID-19 pandemic: is a gender-defined dosage effect responsible for the high mortality rate among males?
Immunogenetics (2020)
-
Toll-like receptors in lupus nephritis
Journal of Biomedical Science (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
