
Abstract
While it is increasingly recognized that three-dimensional (3D) cell culture models recapitulate drug responses of human cancers with more fidelity than monolayer cultures, a lack of quantitative analysis methods limit their implementation for reliable and routine assessment of emerging therapies. Here, we introduce an approach based on computational analysis of fluorescence image data to provide high-content readouts of dose-dependent cytotoxicity, growth inhibition, treatment-induced architectural changes and size-dependent response in 3D tumour models. We demonstrate this approach in adherent 3D ovarian and pancreatic multiwell extracellular matrix tumour overlays subjected to a panel of clinically relevant cytotoxic modalities and appropriately designed controls for reliable quantification of fluorescence signal. This streamlined methodology reads out the high density of information embedded in 3D culture systems, while maintaining a level of speed and efficiency traditionally achieved with global colorimetric reporters in order to facilitate broader implementation of 3D tumour models in therapeutic screening.
Similar content being viewed by others


Introduction
The attrition rates for preclinical development of oncology therapeutics are particularly dismal due to a complex set of factors which includes 1) the failure of pre-clinical models to recapitulate determinants of in vivo treatment response and 2) the limited ability of available assays to extract treatment-specific data integral to the complexities of therapeutic responses1,2,3. Three-dimensional (3D) tumour models have been shown to restore crucial stromal interactions which are missing in the more commonly used 2D cell culture and that influence tumour organization and architecture4,5,6,7,8, as well as therapeutic response9,10, multicellular resistance (MCR)11,12, drug penetration13,14, hypoxia15,16 and anti-apoptotic signaling17. However, such sophisticated models can only have an impact on therapeutic guidance if they are accompanied by robust quantitative assays, not only for cell viability but also for providing mechanistic insights related to the outcomes. While numerous assays for drug discovery exist18, they are generally not developed for use in 3D systems and are often inherently unsuitable. For example, colorimetric conversion products have been noted to bind to extracellular matrix (ECM)19 and traditional colorimetric cytotoxicity assays reduce treatment response to a single number reflecting a biochemical event that has been equated to cell viability (e.g. tetrazolium salt conversion20). Such approaches fail to provide insight into the spatial patterns of response within colonies, morphological or structural effects of drug response, or how overall culture viability may be obscuring the status of sub-populations that are resistant or partially responsive. Hence, the full benefit of implementing 3D tumour models in therapeutic development has yet to be realized for lack of analytical methods that describe the very aspects of treatment outcome that these systems restore.
Motivated by these factors, we introduce a new platform for quantitative in situ treatment assessment (qVISTA) in 3D tumour models based on computational analysis of information-dense biological image datasets (bioimage-informatics)21,22. This methodology provides software end-users with multiple levels of complexity in output content, from rapidly-interpreted dose response relationships to higher content quantitative insights into treatment-dependent architectural changes, spatial patterns of cytotoxicity within fields of multicellular structures and statistical analysis of nodule-by-nodule size-dependent viability. The approach introduced here is cognizant of tradeoffs between optical resolution, data sampling (statistics), depth of field and widespread usability (instrumentation requirement). Specifically, it is optimized for interpretation of fluorescent signals for disease-specific 3D tumour micronodules that are sufficiently small that thousands can be imaged simultaneously with little or no optical bias from widefield integration of signal along the optical axis of each object. At the core of our methodology is the premise that the copious numerical readouts gleaned from segmentation and interpretation of fluorescence signals in these image datasets can be converted into usable information to classify treatment effects comprehensively, without sacrificing the throughput of traditional screening approaches. It is hoped that this comprehensive treatment-assessment methodology will have significant impact in facilitating more sophisticated implementation of 3D cell culture models in preclinical screening by providing a level of content and biological relevance impossible with existing assays in monolayer cell culture in order to focus therapeutic targets and strategies before costly and tedious testing in animal models.
Using two different cell lines and as depicted in Figure 1, we adopt an ECM overlay method pioneered originally for 3D breast cancer models23 and developed in previous studies by us to model micrometastatic ovarian cancer19,24. This system leads to the formation of adherent multicellular 3D acini in approximately the same focal plane atop a laminin-rich ECM bed, implemented here in glass-bottom multiwell imaging plates for automated microscopy. The 3D nodules resultant from restoration of ECM signaling5,8, are heterogeneous in size24, in contrast to other 3D spheroid methods, such as rotary or hanging drop cultures10, in which cells are driven to aggregate into uniformly sized spheroids due to lack of an appropriate substrate to adhere to. Although the latter processes are also biologically relevant, it is the adherent tumour populations characteristic of advanced metastatic disease that are more likely to be managed with medical oncology, which are the focus of therapeutic evaluation herein. The heterogeneity in 3D structures formed via ECM overlay is validated here by endoscopic imaging of in vivo tumours in orthotopic xenografts derived from the same cells (OVCAR-5).

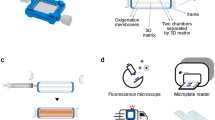
A simplified schematic flow chart of imaging-based quantitative in situ treatment assessment (qVISTA) in 3D cell culture.
(This figure was prepared in Adobe Illustrator® software by MD Glidden, JP Celli and I Rizvi). A detailed breakdown of the image processing (Step 4) is provided in Supplemental Figure 1.
We utilize the methods introduced here to quantify patterns of cytotoxic response in two disease systems, 3D ovarian and pancreatic tumour models, subject to a panel of five clinically-relevant cytotoxic interventions. These treatments represent contrasting mechanisms of action and as shown here, varying patterns of size-dependent, structural, heterogeneous induction of cell death and/or growth inhibition. Carboplatin is a platinum-based agent which coordinates permanently to DNA25, paclitaxel acts on cytoskeletal tubulin to induce defects in mitotic spindle assembly26,27, etoposide is a topoisomerase II inhibitor which induces DNA strand breakage by preventing re-ligation28, gemcitabine is a pyrimidine nucleoside analog which interrupts cell division by replacing cytidine during DNA replication29 and PDT, with the photosensitizer verteporfin, induces apoptosis by localized generation of singlet oxygen to damage anti-apoptotic Bcl-family proteins and trigger direct release of cytochrome c30,31. Therapeutic agents are probed in disease models relevant to their clinical implementation. For example, response to carboplatin and paclitaxel, used in clinical management of advanced ovarian cancer32 are examined in a 3D ovarian tumour model, while gemcitabine, which is the standard agent for palliative care of metastatic pancreatic cancer33, is examined in 3D culture models of that disease. In the former, the importance of providing a systematic approach to analyse size-dependent response is underscored by clinical reports that the size of residual disease is one of the most important prognostic indicators of treatment outcome for this often-lethal gynecologic malignancy34,35,36,37. But in addition to demonstrating the utility of the qVISTA methodology, the high-content analysis of response undertaken here could have significance in providing correlations between cell death and tumour structure. We present the case that elucidating the relationship between cell death and tissue organization may also provide a powerful tool to identify synergistic combinations of therapies analyzed herein, such as carboplatin/paclitaxel32 and PDT/carboplatin19 combinations for treatment of advanced ovarian cancer.
Results
Rescaling of vital dye fluorescent signals to provide a ratiometric dose-response metric
We first examined overall dose response to classic cytotoxic drugs in 3D ovarian micronodules to establish a viability metric combining the signals from two fluorescent reporters. Cultures prepared by overlay of OVCAR5 cells on growth factor reduced (GFR) Matrigel in multiwell plates were allowed to form heterogeneous 3D acini consisting of tens to hundreds of cells, as previously characterized19,24.
Cultures were assigned into control (no treatment, NT and total killing, TK) and treatment groups for administration of paclitaxel, carboplatin, etoposide or PDT. In Figure 2a, representative merged multichannel images of calcein and ethidium bromide stained cultures (green and red respectively) obtained at low magnification are shown for a range of paclitaxel doses. Images from each well are obtained using an automated scanning stage. On a laser scanning confocal system one can obtain 96 spatial fields (at 512 by 512 resolution), with 3 channels per field (1 brightfield and 2 fluorescence channels) in approximately 25 minutes, or less than half this time for widefield illumination imaged onto a CCD chip. In this manner, large fluorescence image sets containing spatially resolved images of esterase activity (cleavage of calcein AM) and membrane permeability (intercalation of ethidium bromide), in thousands of labeled nodules for each treatment condition, are rapidly obtained. Average background-corrected esterase activity in 3D cultures decreases with increasing cytotoxic doses while the membrane permeabilization increases, as expected (Figure 2b).

A viability metric reports overall dose response and heterogeneity based on relative esterase activity and membrane permeability.
In (a), representative thumbnails from merged images of esterase-cleaved calcein AM (green) and intercalated ethidium bromide (red) in ovarian cancer 3D overlays for (left to right), no treatment (NT) and total killing (TK) and increasing doses of paclitaxel (as a representative cytotoxic agent). Thumbnails qualitatively display increasing ethidium bromide intercalation (red) and decreasing esterase activity (green) with increasing cytotoxic dose similar for all therapies assayed here. Scale bars = 500 μm. In (b), mean fluorescence signals from full fields after rescaling (Equation 1) are shown for escalating doses of paclitaxel, carboplatin, etoposide and verteporfin PDT treatments (from left to right) in the upper plots used to quantify overall viability (lower plots, from Equation 2) and estimate fractional lethal doses (LD). In (c), distributions of nodule viabilities (N > 1000) from automated segmentation (shown for paclitaxel-treated cultures) shifts to the left as expected with increasing dose. In (d), paclitaxel and carboplatin viability distributions at LD80 (thumbnails, left, scalebars = 500 μm) are juxtaposed to reveal a relatively tight distribution in the former in contrast to heterogeneous viability spectrum in the latter with sub-populations up to 80–90% viable.
To correct for incompatible baseline fluorescent signals and scaling from separate reporters, a scaling factor, ϕ, which establishes internally consistent “live” and “dead” signals is based on the NT and TK controls for each experiment. The TK control (fixed and permeabilized), establishes a lower limit of esterase activity with a high degree ethidium bromide intercalation, in otherwise identically prepared cultures and imaging conditions, (analogous to a “blank” group). The values of ϕ for the data shown in Figure 2b are 1.09, 1.04, 2.39 and 1.13 for paclitaxel, carboplatin, etoposide and verteporfin PDT, respectively. The rescaled values (Figure 2b, lower panels) are then appropriately combined into a single metric where viability is interpreted as the quotient of live to total fluorescent signal (the sum of live and rescaled dead signals). Even with this effort to rescale in a manner is not therapeutic-dependent, the dynamic range of the signal from intercalated ethidium bromide may vary due to treatment dependent effects. For example the ethidium bromide signal at exceptionally high carboplatin doses is higher than that of the TK control possibly due to dye binding or optical effects of treatment-induced DNA damage, hence the rescaled calcein signal appears relative flat when plotted on the same scale as in Figure 2B even though it decreases significantly in a dose-dependent manner. Overall this metric provides a simple and rapid means for reporting fractional lethal doses (LD), which is validated here by counting of trypan blue excluding cells (Supplemental Figure 2).
As an imaging-based approach, this methodology also generates viability statistics for all individual nodules imaged in each set of multichannel images. The viability readout is mapped to each segmented region (nodule) to quantify heterogeneity in treatment response. As shown in Figure 2c, with increasing cytotoxic dose, the distribution of viabilities shifts to the left as expected with increasing cell death. For the data shown here for paclitaxel-treated cultures the viability distribution for each dose is relatively compact and fits to a single Gaussian mode with a standard deviation of 0.11 at the LD80 dose. When the viability distributions for equivalent LD80 doses of paclitaxel and carboplatin are overlaid (Figure 2d) it is clear that there is a broader viability spectrum exhibited by the latter. With a wide distribution of viability to 80–90% viable and a small shoulder of completely killed nodules (0–10% viable) at the left side of the distribution the carboplatin distribution has a far more heterogeneous response with a width (standard deviation) of 0.20.
High-content quantification of viabilities in large distributions of individual micronodules reports differential size-dependent response
To establish that heterogeneity in nodule size is reflective of in vivo growth behavior, the same cells were implanted into mice, as previously described for a murine ovarian xenograft model38 and imaged using a high-resolution fluorescence microendoscopy approach previously established by us39. As shown in Figure 3, the bi-modal size distribution of nodules overlaid on extracellular matrix resembles the heterogeneity in size of nodular implants on peritoneal surfaces in vivo. In 3D culture data, to co-register size and cytotoxic response data, segmented regions are mapped to readouts of size, reported as volume (V) from an ellipsoid approximation, estimated number of cells (N), equivalent diameter (deq), major and minor axis lengths, eccentricity (e) and spatial position (x and y- coordinates) in addition to calcein and scaled ethidium bromide fluorescence signals and viability, of each nodule in each field for each treatment group, totaling several thousand for each multi-well plate imaged. A truncated representation of these readouts is shown for a small sub-region of a single paclitaxel-treated well in a 96-well plate is shown in Figure 3b. From this larger numerical dataset, size-viability scatter plots are shown in Figure 3c, in which each data point indicates the volume (in μm3, horizontal axis) and viability (vertical axis) for a given nodule. In general, the densest regions of the scatter shift toward lower positions on the vertical axis as expected with increasing cytotoxic dose and consistent with the overall average values (for non-segmented full image fields) in Figure 2. While paclitaxel therapy induces cytotoxicity nearly uniformly over nodule size, carboplatin and etoposide, even at extremely high doses, show a significant proportion of larger nodules that remain viable with, especially for carboplatin, a dense clustering of completely non-viable nodules in the smallest size range. This suggests a size-dependent response in which smaller nodules are more sensitive to treatment than larger nodules, which is typical of clinical treatment response37. The large spread in viability of nodules exposed to etoposide across all sizes at the ~LD50 dose suggests that there is a high degree of heterogeneity with populations resistant to this agent. PDT treatment using the photosensitizer verteporfin also exhibits a high degree of heterogeneity, manifested in particular by the large vertical smear of points at small sizes (less than ~10 cells), with a size-dependent trend for larger nodules.

Size heterogeneity in vitro and in vivo and size-dependent therapeutic response.
(a): In vivo nodule size heterogeneity from representative fluorescent microendoscope fields of xenograft implanatation of ovarian cancer cells, which resembles size distribution of the same cells grown in vitro. Endoscope fields are 800 μm in diameter. (b): A representation of our use of segmentation in 3D culture image data to identify individual nodules from the brightfield channel to generate a mask for computation of multiple readouts (shown in truncated form) for each individual nodule. In (c), viability-volume scatter plots output by qVISTA code showing the rescaled viability of each nodule with respect to its volume, for thousands of nodules in each treatment group, for each treatment. The vertical spread reflects dose response heterogeneity and for given a range of sizes. Probing size-dependent response in this manner reveals, for example, that the only nodules that are completely responsive to carboplatin are the smallest volume nodules while the largest remain weakly viable even at a 1000 μM (LD80) dose. Response to paclitaxel is the most homogeneous and size-independent of therapies evaluated. Etoposide-treated cultures exhibit a broad spread of viabilities over a broad spread of nodule sizes, while verteporfin-PDT treatment induces a pronounced bi-modal response with simultaneously emergent populations of small nodules (1–10 cells) that are almost completely viable and almost completely non-viable.
To verify that quantification of size-dependent effects are not skewed by optical bias at the relevant wavelengths, we evaluated the scaling relationship between the integrated fluorescence signal (for each channel from each nodule) versus nodule volume (Supplemental Figure 3). The linear scaling over the majority of data (>85% for the calcein channel) indicates that signal attenuation due to tissue optics effects become significant only for the very largest nodules, which are also the most problematic for volume estimation by the ellipsoid approximation applied here. Here we examine these effects in the calcein channel, which requires shorter wavelength excitation that is more highly scattered (relative to the ethidium bromide excitation light). Importantly, the validity of fluorescence quantification over a wide range of nodule sizes is further established by the strong agreement between the average viability calculated from fluorescence and manual counting of cells (Supplemental Figure 2).
Quantification of architectural changes following cytotoxic intervention reveals treatment and dose-dependent nodular disruption
The ability to place treatment response in the context of nodule size raises the question of the extent to which treatments induce changes in the size distribution by breakup or “unpacking” of multicellular nodules. In high resolution confocal images of 3D nodules stained with vital dyes following carboplatin or verteporfin photodynamic therapy (PDT), in contrast to no treatment controls (Figure 4a), the presence of a number of detached single cells around the periphery of a large nodular core suggest that the latter therapy causes large (>200 μm) multicellular nodules to disassociate and break down into smaller clusters of cells. Informed by a previous characterization study24, 3D micronodules reproducibly exhibit distinct log-normal size modes described by a bimodal Gaussian f = f1 + f2. We quantify the extent to which treatment-induced disruption causes shifts in the proportion of nodules in the mode corresponding to larger sizes (f2) to smaller sizes (f1). As seen in Figure 4b, there is a marked dichotomy in the ability of different therapeutics to induce this size-distribution shift, with pronounced effects from vertporfin-PDT and paclitaxel treatments, but no shift toward smaller sizes following etoposide or carboplatin treatments. We describe the extent of this shift as disruption fraction, or Dfrac, a value which ranges from −1 to 1. In (Figure 4c), dose-dependent Dfrac is reported relative to the extent of cytotoxic lethality for selected doses (LD20, LD50, etc) of each agent. Dfrac increases in a sharply dose-dependent manner for verteporfin PDT treatments, while carboplatin treatment suppresses viability at very high doses with no significant architectural disruption. Etoposide treatment displays a flat Dfrac curve, while paclitaxel induces a marked nodular disaggregation, it is seen only at higher lethal doses relative to PDT. A byproduct of the definition of Dfrac, is that negative values indicate a higher proportion of large (non-disrupted) nodules in treated, relative to untreated, cultures. As seen in immunofluorescence images (Figure 4d) there is a clear contrast between strong E-cadherin staining at cell junctions in untreated nodules, with disrupted punctate intracellular E-cadherin staining in PDT-treated nodules, consistent with the observation of disruption and decreased cell-cell contact.

Contrasting quantitative reporters of cytotoxic and structural response.
In (a) high-resolution 3D confocal renderings of single nodules from no treatment, carboplatin and verteporfin-PDT are shown from top to bottom to provide qualitative assessment of contrasting patterns of cytotoxic response. Scale bars = 50 μm. In (b), log-normal bi-modal nodule volume distributions show contrasting patterns between with little evidence of disruption (upper plots, given doses of etoposide and carboplatin) and treatments which with appropriate dosage appear to have a propensity to “unpack” multicellular nodules (lower plots, paclitaxel and PDT). Each histogram contains statistics on 1,000–3,000 micronodules. In the latter category there is a shift in the bimodal size distribution from the proportion of nodules in the mode corresponding to larger sizes to smaller sizes consistent with larger nodules being induced to disaggregate into an increased number of small nodules. In (c), disruption fraction, (Dfrac) based on the treatment-induced shifts in size from (B), is reported with respect to the degree of lethality for several dose for each treatment tested. Error bars show standard error. Verteporfin-PDT induces high Dfrac at low lethality while carboplatin and etoposide treatments suppresses viability at very high doses with negligible architectural disruption. Paclitaxel also unpacks nodules but requires higher level of cell killing in order to do so. In (d), immunofluorescence images show a clear contrast between strong E-cadherin staining at cadherens junctions in untreated nodules, with disrupted and punctate intracellular staining in PDT-treated nodules, consistent with the observation of cellular disassociation.
Quantification of growth inhibitory and cytotoxic treatment effects
To examine a scenario in which it is useful to differentiate cytotoxic from growth-inhibitory effects, we studied the contrasting response of 3D pancreatic tumour nodules to gemcitabine and paclitaxel. Visualization of gemcitabine response from microscope fields stained with vital dyes (Figure 5a) shows that there is little or no evidence of cell death, even at high concentrations. This is reflected in a nearly flat dose response curve as reported by the intensity-based viability metric (Figure 5c). However, summation of the estimated total volume of residual viable nodules overlaid on the same plot (Figure 5b) reveals that there is a modest inhibition of growth. In pancreatic 3D cultures treated with paclitaxel, we see a pronounced cytotoxic response reflected in the image data (Figure 5b) and reported by both treatment response metrics (Figure 5d). The similarity in paclitaxel response with ovarian 3D cultures (Figures 2 and 3) indicates that the insensitivity to gemcitabine at nearly 100 times the monolayer LD50 dose40 is reflective of the contrasting action of the therapy, rather than the disease model. A slight disagreement between intensity-based and segmentation-based (volume) metrics at high cytotoxic doses (~90% killing) reflects segmentation error in processing images with such weak calcein signal relative to background. The intensity-based viability metric, based on ratiometric scaling of two fluorescent signals is however in close agreement with validation by trypan blue exclusion counts (Supplemental Figure 2) even at these high doses, making it a more appropriate metric to quantify the action of a highly cytotoxic agent such as paclitaxel.

Analysis of contrasting growth inhibitory and cytotoxic responses in a 3D pancreatic tumour model.
(a): Representative thumbnails from merged fluorescent images of cleaved calcein AM (green) and intercalated ethidium bromide (red) for pancreatic 3D cultures treated with (from left to right) untreated, 0.01, 1.0 and 100 μM gemcitabine doses showing minimal evidence of cell death (ethidium bromide staining). (b): Analogous thumbnails for identical pancreatic 3D cultures (left to right) untreated, or treated with 2.78, 11.1 or 44.5 μM paclitaxel dosage. In (c), estimation of tumour volumes reports a modest growth inhibition (~20%) from gemcitabine treatment which is not detected by the intensity-based viability metric. In (d), a strong cytotoxic response to paclitaxel is reported by both metrics, with some disagreement from at doses nearing 90% killing due to error in segmentation of weak calcein signals in those treated cultures.
Discussion
We introduce a robust methodology to provide rapid readouts of multiple treatment assessment endpoints from 3D in vitro tumour models. This system not only overcomes technical barriers confounding the use of standard colorimetric assays in this context, but also provides a degree of information content not approached by such assays. We demonstrate the value of new quantitative descriptors of therapeutic response for five different oncology agents. The therapeutics evaluated here, while primarily driving cell death through apoptosis, do so through contrasting molecular mechanisms with, as shown in this study, markedly contrasting patterns of structural and size-dependent response, heterogeneous induction of cell death and/or growth inhibition, as summarized in Table 1. The characteristic fingerprints of therapeutic behavior summarized in ??? can be plausibly connected with more general phenomena, such as drug resistance and drug penetration and/or the collective behavior of multicellular nodules exposed to a given stressor. For example, the former is suggested by the profound heterogeneity observed in response to etoposide treatment. Given this non-uniformity of response, for an LD50 dose of this agent there are significant fractions of nodules that remain completely viable while others that are completely killed. In contrast to paclitaxel for example, with a nearly uniform suppression of viability, this suggests that each multicellular nodule is 50% dead, consisting of nearly 50% dead cells (outer layers) and 50% viable cells (an inner core). It is also worth noting that while paclitaxel treatment can drive essentially all cells in culture to nearly absolute zero esterase activity at high doses, cultures subject to carboplatin exhibit populations of cells that remain weakly esterase positive even with extremely high dosage thus exhibiting a lower viability plateau of approximately 10% (Figure 1b). This reduction of metabolic activity concomitant with persistent survival could be a manifestation of a reversible senescent state, as recently shown for cisplatin treatment, a chemically analogous agent41. These quantitative insights are reported rapidly using the combination of 3D tumour models and high-content image analysis described here.
We introduce a metric, Dfrac, which quantifies the extent of disruption and fragmentation of multicellular nodules that results from treatment with a particular agent. In contrasting the marked difference in nodular disruption relative to cytotoxicity for carboplatin and verteporfin-based PDT treatments examined here, it is provocative that this new quantitative vocabulary provides a basis to account for the previously reported synergistic enhancement in carboplatin efficacy when pre-treated with PDT19. The concept that this enhancement results in part from an increase in carboplatin penetration following pre-treatment with PDT is supported by the high degree of nodular disruption induced by the latter. The poor penetration of carboplatin into large multicellular nodules, perhaps resulting from the inability to unpack, or disaggregate tumours, is further supported by the observed size-dependent response of this agent reported here37. Importantly, the ability to quantify these treatment-induced changes suggests the ability to utilize this approach for predictive identification of combination therapies. For example, therapies that are identified to disrupt tumour architecture (high Dfrac) could be indexed for combination with those that exhibit weak penetration through tight cell junctions yet are otherwise effective. It is provocative that paclitaxel and carboplatin, which are used in combination as first line therapy for management of advanced ovarian cancer32, are revealed here to have inversely related structural effects. The nearly equal and opposite levels of architectural disruption (where Dfrac is negative for carboplatin) seen at an LD90 level for these two agents indicates that the weakly viable micronodules left behind are relatively small in one case and relatively large in the other case. This could contribute to a mechanistic explanation of how these agents in combination could achieve a more complete response at safer doses. The ability to make such correlations between structural and cytotoxic responses could be invaluable in screening and informing rationally-designed combinations of the next generation of oncology agents.
A critical component of the imaging-based strategy introduced here is the rational tradeoff of image-acquisition parameters for field of view, depth of field and optical resolution and the development of image processing routines for appropriate removal of background, scaling of fluorescence signals from more than one channel and reliable segmentation of nodules. In order to obtain depth-resolved 3D structures for each nodule at sub-micron lateral resolution using a laser-scanning confocal system, it would require ~ 40 hours (at approximately 100 fields for each well with a 20× objective, times 1 minute/field for a coarse z-stack, times 24 wells) to image a single plate with the same coverage achieved in this study. Even if the resources were available to devote to such time-intensive image acquisition, not to mention the processing, the optical properties of the fluorophores would change during the required time frame for image acquisition, even with environmental controls to maintain culture viability during such extended imaging. The approach developed here, with a mind toward adaptation into high throughput screening, provides a rational balance of speed, requiring less than 30 minutes/plate and statistical rigour, providing images of thousands of nodules in this time, as required for the high-content analysis developed in this study. These parameters can be further optimized for specific scenarios. For example, we obtain the same number of images in a 96 well plate as for a 24 well plate by acquiring only a single field from each well, rather than 4 stitched fields. This quadruples the number conditions assayed in a single run, at the expense of the number of nodules per condition and therefore the ability to obtain statistical data sets for size-dependent response, Dfrac and other segmentation-dependent numerical readouts.
We envision that the system for high-content interrogation of therapeutic response in 3D cell culture could have widespread impact in multiple arenas from basic research to large scale drug development campaigns. As such, the treatment assessment methodology presented here does not require extraordinary optical instrumentation or computational resources, making it widely accessible to any research laboratory with an inverted fluorescence microscope and modestly equipped personal computer. And although we have focused here on cancer models, the methodology is broadly applicable to quantitative evaluation of other tissue models in regenerative medicine and tissue engineering. While this analysis toolbox could have impact in facilitating the implementation of in vitro 3D models in preclinical treatment evaluation in smaller academic laboratories, it could also be adopted as part of the screening pipeline in large pharma settings. With the implementation of appropriate temperature controls to handle basement membranes in current robotic liquid handling systems, our analyses could be used in ultra high-throughput screening. In addition to removing non-efficacious potential candidate drugs earlier in the pipeline, this approach could also yield the additional economic advantage of minimizing the use of costly time-intensive animal models through better estimates of dose range, sequence and schedule for combination regimens.
Methods
Three-dimensional cell culture
NIH:OVCAR-5 epithelial ovarian cancer cells were obtained from Thomas Hamilton (Fox Chase Cancer Institute, Philadelphia, PA). PANC-1 cells were obtained from American Type Culture Collection (ATCC, Rockville, MD) and maintained according to ATCC instructions. Cell media and fetal bovine serum were obtained from Mediatech (Herndon, VA) and Invitrogen (Carlsbad, CA), respectively. Cells were maintained in either RPMI-1640 medium (OVCAR-5) or DMEM (PANC-1) supplemented with 10% heat-inactivated fetal calf serum, 50 U/mL penicillin and 50 μg/mL streptomycin. 3D cultures were prepared in overlay geometry adapting a method previously described by Lee et al23. Briefly, a 1 mL volume of single cell suspension of 7,500 cells/mL in complete medium containing 2% Growth Factor Reduced (GFR) Matrigel was overlaid on beds of GFR Matrigel on glass-bottom 24-well plates (Greiner BioOne). GFR Matrigel beds were initially prepared by ejecting 250 μL of Matrigel solution at ~4°C on chilled culture plates and carefully rotating at a slight angle to distribute evenly. This method forms a bed that is approximately 500 μm thick near the center of the well where images were obtained (the ECM bed is thicker near the edges due to a meniscus effect). Cultures were then incubated at 37°C for 30 minutes prior to plating cells to allow gelation to occur. For growth on 96 well plates, the cell count was scaled down to 1250 cells in 100 μL media to account for a growth area that is approximately one sixth that of a 24 well plate with a reduction in GFR Matrigel bed volume in the same ratio. Following initial dispensing, cells were allowed to adhere to the Matrigel bed before addition of complete growth medium containing 2% GFR Matrigel. As noted previously23, the highly diluted 2% GFR Matrigel supplementing the complete growth medium in the overlay does not form a gel, but does provide ECM signaling cues throughout culture growth for cells not in physical contact with the underlying ECM bed. All cultures were maintained in an incubator at 37°C in an atmosphere of 5% CO2.
Chemotherapy treatments
Chemotherapy treatments were performed by incubating 3D cultures in growth medium with the indicated dosage of carboplatin (NDC: 61703-360-22; NOVAPLUS, Irving, TX, USA) diluted to 5–2560 μM, paclitaxel (NDC: 61703-342-22; Hospira Worldwide, Inc. Lake Forest, IL, USA), etoposide (NDC: 0703-5653-01; Teva Pharmaceuticals, Irvine, CA, USA) diluted to 1–100 μM, or gemcitabine (NDC: 0781-3282-75; Eli Lily and Company, Indianapolis, IN, USA) diluted to 0.01 to 1000 μM. Chemotherapy agents were allowed to incubate for 72 hours with the exception of caroboplatin incubations which were conducted for a total of 96 hours with a refresh at 48 hours. The incubation times chosen for this study are consistent with previous reports and reflect the observation that 3D tumor models typically require a longer period of drug exposure relative to traditional monolayer cultures and these treatment schedules more closely resemble in vivo therapeutic assessment scenarios19,40,42. All chemotherapy drugs were clinical grade formulations purchased from the central pharmacy at the Massachusetts General Hospital. Treatment response was assessed immediately after removal of the chemotherapeutic agent.
In vivo high-resolution fluorescence microendoscopy
For in vivo comparisons we used a murine model of disseminated ovarian carcinoma previously established in our laboratory38. Briefly, female Swiss nude mice (Cox, Cambridge, MA), 6–8 weeks old, were injected intraperitoneally 3.15 × 107 OVCAR5 cells in single cell suspension in 2 mL PBS. Tumours were allowed to develop for 14 days prior to imaging, during which time mice had continual access to food and water while being housed in laminar flow racks under specific pathogen-free conditions. Microendoscopy was performed as previously described39, using intraperitoneal injection with 1 mg/kg body weight of verteporfin (benzoporhyrin derivative monoacid ring-A or BPD-MA, VWR, Radnor, PA, USA) for fluorescence contrast. Imaging was performed under anesthesia with the fibre probe introduced into the intraperitoneal cavity via a catheter inserted through the abdominal wall. Approximately 20 separate microendoscope fields were obtained throughout the peritoneal cavity for each mouse. Microendoscope images were segmented in the same manner as described below for 3D cell cultures to obtain the distribution of tumour sizes throughout the peritoneal cavity. All animal procedures were conducted in accordance with institutional guidelines for research animal care.
Photodynamic therapy treatments
Cell cultures were incubated with medium containing 1 μM verteporfin for 90 minutes prior to irradiation, at which time the medium was replaced with verteporfin-free medium. Cells were irradiated at specified doses using a 690 nm laser diode source (Model 7401; Intense, North Brunswick, NJ) at a fluence rate of 150 mW/cm2 as measured via a VEGA laser power energy meter (Ophir Laser Measurement Group, LLC) and then allowed to incubate for 24 hours prior to evaluation of treatment response. Laser irradiation was delivered through the bottom of each well via a vertically mounted Thorlabs multimode fiber (FT600EMT, Thorlabs, Newton, NJ), collimated to overfill the dish area for nearly uniform light delivery.
Confocal immunofluorescence imaging
Preparation of 3D culture for immunofluorescence imaging was adapted from a procedure previously described by Debnath et al43. Cultures were initially prepared in glass bottom 24 well plates as described above and grown for the specified duration prior to paraformaldehyde fixation (2%) and permeabilization with 0.5% Triton X-100. The exchange of buffers and staining solutions was accomplished using a micropipetter to minimize disruption of the Matrigel. The anti-E-cadherin primary antibody used in this study was conjugated directly to Alexa Fluor 488 (Cell Signaling, Danvers, MA, USA). After incubation with antibody solution, cells were washed and covered with mounting medium containing DAPI (Prolong Antifade with DAPI, Invitrogen, Carlsbad, CA) to label nuclei. All confocal immunofluorescence images were obtained using a 40× water immersion objective, with pinhole open to 180 μm, on an Olympus FV-1000 confocal microscope (Olympus of America) and configured for simultaneous imaging of DAPI and Alexa Fluor 488 fluorescence emission without spectral bleed through or cross talk between channels.
Fluorescence Imaging
Live cultures were stained, in situ, with Calcein AM (Invitrogen) and Ethidium Bromide (Sigma Aldrich) as fluorescent markers for live and dead cells respectively and returned to the incubator for 40 minutes prior to imaging. For wells designated as total killing (TK) controls, the additional steps of fixation and permeabilization were performed prior to addition of fluorescence reagents, to establish control conditions for maximal ethidium bromide intercalation. In this case, cultures were fixed and permeabilized as for samples prepared for immunofluorescence (see above). No treatment (NT) groups within each batch were grown and maintained identically to treated groups and subject to sham manipulations prior to imaging-based evaluation at the same time as other treatment groups. All images were obtained using an Olympus FV-1000 confocal microscope using a 0.16NA 4× objective to obtain stitched multichannel mosaics of 4 512 × 512 pixel image fields spanning 6.3 mm × 6.3 mm from the central region of each well. Images were acquired in a de facto widefield limit, with the pinhole open to 600 um, so as to allow for maximum integration of fluorescence emission over the z-dimension of 3D objects without the high time cost of refocusing and acquiring stacks at multiple z-positions for each spatial field during this time-sensitive procedure. The 488 nm line from an Argon ion laser and a 559 nm laser diode were used for excitation of the converted calcein, imaged through a FITC emission filter and intercalated ethidium bromide, imaged through a TRITC emission filter, respectively. Saved images were grouped into a pre-assigned system of subdirectories for off-line batch-processing in MATLAB.
qVISTA image processing routines
The ratiometric measure of overall mean culture viability is obtained from spatially averaged intensities of calcein (CG, live) and ethidium bromide (EtBr, dead) fluorescence signals (after background subtraction) in image fields for each treatment group as shown in Equation 1:

The subscripts NT and TK designate mean values for no-treatment (NT) and total-killing (TK) controls. The scaling factor ϕ rescales all values to internal non-treated and total killing controls and corrects for inevitable variability in imaging parameters and minor instrumentation drift across evaluation timepoints to provide quantitative analysis that corrects for these parameters. This rescaling is critical to introduce standardized intensity “units” that are compatible between two separate fluorescent channels from two fluorophores, excited with different lasers, imaged through separate optical filters. All fluorescence intensity measurements are obtained from background corrected images, where the image histogram is automatically tabulated to determine mean and mode intensity.
To generate plots of size-dependent treatment response from images containing heterogeneous distributions of multicellular 3D micronodules, multi-resolution wavelet decomposition44 of brightfield images was first used for gradient background removal as shown in Supplemental Figure 1. In order to generate a mask for subsequent processing, background-corrected images were segmented, using a built in MATLAB routine based on an implementation of Otsu's method, as previously described for similar image data24. The volume of each individual segemented object was calculated based on a best-fit ellipsoid of major and minor axis a and b respectively (Equation 2). Other parameters, including eccentricity (second moment estimate), centroid coordinates and equivalent (circular) diameter are automatically tabulated and reported for each nodule. The segmented image was also used to generate a binary mask, which was multiplied by each fluorescent channel to obtain viability for each masked region (each micronodule) using the same process for rescaling and internally normalizing viabilities as described above. In this manner, viability-volume scatter plots were generated. Additionally, the same segmentation procedure was invoked on the esterase-cleaved calcein image channels for each treatment group to obtain statistics on the size (volume) distribution of residual calcein positive (viable) nodules or regions thereof. Summation over all nodule volumes (Vn) in each group provides an estimate of fractional residual volume relative to the NT group, VNT, which serves as an additional metric describing overall cytotoxic response:

For quantification of architectural disruption in response to treatment we fit to the to the previously reported bimodal lognormal size distribution that describes in vitro nodule formation in the overlay geometry45 as shown in Equation 3:

Upon treatment the size distribution of residual viable disease may shift, (Figure 4) as larger nodules into smaller fractions. To quantify this observation we introduce a metric, disruption fraction (Dfrac) which reports the fractional shift from the larger size mode (f2) to the smaller mode (f1) in size distributions relative to NT controls:

In practice, for economy of computation speed and to avoid instabilities from repeated non-linear least squares curve fits, this calculation is performed as the equivalent summation over discrete volume bins where a cutoff-bin is determined based on the inflection point in the bimodal curve (corresponding to f1 = f2).
Validation of cell viability measurements
For validation of the overall viability metric introduced here, treatment response was also assessed by trypan blue exclusion. Comparisons are provided with the substantial caveat that the lack of suitable assays for assessment of 3D micronodules adherent to ECM and challenges of reliable nodule disaggregation are in fact motivating forces for developing an imaging-based in situ assessment in the present study. In order to obtain reliable cell counts, 3D micronodules from each treatment condition had to first be removed from GFR Matrigel by 2 hour digestion with dispase (BD Biosciences, Bedford, MA, USA), followed by an additional resuspension in 0.25% Trypsin EDTA (Mediatech, Herndon, VA, USA) to disaggregate multicellular nodules. For cell counting experiments, disaggregated cells were then stained with trypan blue before transfer to a hemacytometer.
References
Ocana, A., Pandiella, A., Siu, L. L. & Tannock, I. F. Preclinical development of molecular-targeted agents for cancer. Nat Rev Clin Oncol 8, 200–209 (2011).
Yap, T. A., Sandhu, S. K., Workman, P. & de Bono, J. S. Envisioning the future of early anticancer drug development. Nat Rev Cancer 10, 514–523 (2010).
Sharma, S. V., Haber, D. A. & Settleman, J. Cell line-based platforms to evaluate the therapeutic efficacy of candidate anticancer agents. Nat Rev Cancer (2010).
Bissell, M. J. et al. Tissue structure, nuclear organization and gene expression in normal and malignant breast. Cancer Res 59, 1757–1763s; discussion 1763s–1764s (1999).
Bissell, M. J. & Radisky, D. Putting tumours in context. Nat Rev Cancer 1, 46–54 (2001).
Nelson, C. M. & Bissell, M. J. Of extracellular matrix, scaffolds and signaling: tissue architecture regulates development, homeostasis and cancer. Annu Rev Cell Dev Biol 22, 287–309 (2006).
Muthuswamy, S. K., Li, D., Lelievre, S., Bissell, M. J. & Brugge, J. S. ErbB2, but not ErbB1, reinitiates proliferation and induces luminal repopulation in epithelial acini. Nat Cell Biol 3, 785–792 (2001).
Debnath, J. et al. The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell 111, 29–40 (2002).
Vinci, M. et al. Advances in establishment and analysis of three-dimensional tumor spheroid-based functional assays for target validation and drug evaluation. BMC Biol 10, 29 (2012).
Friedrich, J., Seidel, C., Ebner, R. & Kunz-Schughart, L. A. Spheroid-based drug screen: considerations and practical approach. Nat Protoc 4, 309–324 (2009).
Kobayashi, H. et al. Acquired multicellular-mediated resistance to alkylating agents in cancer. Proc Natl Acad Sci U S A 90, 3294–3298 (1993).
Frankel, A., Buckman, R. & Kerbel, R. S. Abrogation of taxol-induced G2-M arrest and apoptosis in human ovarian cancer cells grown as multicellular tumor spheroids. Cancer Res 57, 2388–2393 (1997).
Minchinton, A. I. & Tannock, I. F. Drug penetration in solid tumours. Nat Rev Cancer 6, 583–592 (2006).
Au, J. L. et al. Determinants of drug delivery and transport to solid tumors. J Control Release 74, 31–46 (2001).
Shannon, A. M., Bouchier-Hayes, D. J., Condron, C. M. & Toomey, D. Tumour hypoxia, chemotherapeutic resistance and hypoxia-related therapies. Cancer Treat Rev 29, 297–307 (2003).
Evans, C. L. et al. Killing hypoxic cell populations in a 3D tumor model with EtNBS-PDT. PLoS ONE 6, e23434 (2011).
Yang, T. M., Barbone, D., Fennell, D. A. & Broaddus, V. C. Bcl-2 family proteins contribute to apoptotic resistance in lung cancer multicellular spheroids. Am J Respir Cell Mol Biol 41, 14–23 (2009).
Kepp, O., Galluzzi, L., Lipinski, M., Yuan, J. & Kroemer, G. Cell death assays for drug discovery. Nat Rev Drug Discov 10, 221–237 (2011).
Rizvi, I. et al. Synergistic Enhancement of Carboplatin Efficacy with Photodynamic Therapy in a Three-Dimensional Model for Micrometastatic Ovarian Cancer. Cancer Res 70, 9319–9328 (2010).
Berridge, M. V., Herst, P. M. & Tan, A. S. Tetrazolium dyes as tools in cell biology: new insights into their cellular reduction. Biotechnol Annu Rev 11, 127–152 (2005).
Eliceiri, K. W. et al. Biological imaging software tools. Nat Methods 9, 697–710 (2012).
Myers, G. Why bioimage informatics matters. Nat Methods 9, 659–660 (2012).
Lee, G. Y., Kenny, P. A., Lee, E. H. & Bissell, M. J. Three-dimensional culture models of normal and malignant breast epithelial cells. Nat Methods 4, 359–365 (2007).
Celli, J. P., Rizvi, I., Evans, C. L., Abu-Yousif, A. O. & Hasan, T. Quantitative imaging reveals heterogeneous growth dynamics and treatment-dependent residual tumor distributions in a three-dimensional ovarian cancer model. J Biomed Opt 15, 051603–051610 (2010).
Knox, R. J., Friedlos, F., Lydall, D. A. & Roberts, J. J. Mechanism of cytotoxicity of anticancer platinum drugs: evidence that cis-diamminedichloroplatinum(II) and cis-diammine-(1,1-cyclobutanedicarboxylato)platinum(II) differ only in the kinetics of their interaction with DNA. Cancer Res 46, 1972–1979 (1986).
Fuchs, D. A. & Johnson, R. K. Cytologic evidence that taxol, an antineoplastic agent from Taxus brevifolia, acts as a mitotic spindle poison. Cancer Treat Rep 62, 1219–1222 (1978).
Jordan, M. A. & Wilson, L. Microtubules as a target for anticancer drugs. Nat Rev Cancer 4, 253–265 (2004).
Nitiss, J. L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat Rev Cancer 9, 338–350 (2009).
Plunkett, W. et al. Gemcitabine: metabolism, mechanisms of action and self-potentiation. Semin Oncol 22, 3–10 (1995).
Kessel, D. & Castelli, M. Evidence that bcl-2 is the target of three photosensitizers that induce a rapid apoptotic response. Photochem Photobiol 74, 318–322 (2001).
Kessel, D. & Luo, Y. Photodynamic therapy: a mitochondrial inducer of apoptosis. Cell Death Differ 6, 28–35 (1999).
du Bois, A. et al. A Randomized Clinical Trial of Cisplatin/Paclitaxel Versus Carboplatin/Paclitaxel as First-Line Treatment of Ovarian Cancer. J Natl Cancer Inst 95, 1320–1329 (2003).
Burris 3rd, H. et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. Journal of Clinical Oncology 15, 2403 (1997).
Akahira, J. I. et al. Prognostic factors of stage IV epithelial ovarian cancer: a multicenter retrospective study. Gynecol Oncol 81, 398–403 (2001).
Guarneri, V., Piacentini, F., Barbieri, E. & Conte, P. F. Achievements and unmet needs in the management of advanced ovarian cancer. Gynecol Oncol 117, 152–158 (2010).
Hoskins, W. J. et al. The effect of diameter of largest residual disease on survival after primary cytoreductive surgery in patients with suboptimal residual epithelial ovarian carcinoma. Am J Obstet Gynecol 170, 974–979; discussion 979–980 (1994).
Winter 3rd, W. E. et al. Tumor residual after surgical cytoreduction in prediction of clinical outcome in stage IV epithelial ovarian cancer: a Gynecologic Oncology Group Study. J Clin Oncol 26, 83–89 (2008).
Molpus, K. L. et al. Characterization of a xenograft model of human ovarian carcinoma which produces intraperitoneal carcinomatosis and metastases in mice. Int J Cancer 68, 588–595 (1996).
Zhong, W. et al. In vivo high-resolution fluorescence microendoscopy for ovarian cancer detection and treatment monitoring. Br J Cancer 101, 2015–2022 (2009).
Celli, J. P., Solban, N., Liang, A., Pereira, S. P. & Hasan, T. Verteporfin-based photodynamic therapy overcomes gemcitabine insensitivity in a panel of pancreatic cancer cell lines. Lasers Surg Med 43, 565–574 (2011).
Sharma, S. V. et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 141, 69–80 (2010).
Stamelos, V. A., Robinson, E., Redman, C. W. & Richardson, A. Navitoclax augments the activity of carboplatin and paclitaxel combinations in ovarian cancer cells. Gynecol Oncol 128, 377–382 (2013).
Debnath, J. & Brugge, J. S. Modelling glandular epithelial cancers in three-dimensional cultures. Nat Rev Cancer 5, 675–688 (2005).
Mallat, S. G. A Theory for Multiresolution Signal Decomposition: The Wavelet Representation. IEEE Trans Pattern Analysis Machine Intelligence 11, 674–693 (1989).
Chen, X. et al. Integrin alpha1beta1 regulates epidermal growth factor receptor activation by controlling peroxisome proliferator-activated receptor gamma-dependent caveolin-1 expression. Mol Cell Biol 30, 3048–3058.
Acknowledgements
We gratefully acknowledge constructive input from Dr. Bryan Spring, Dr. Adnan Abu-Yousif and Dr. Conor Evans, of the Wellman Center for Photomedicine, arising from discussions about the image analysis routines described herein. Dr. Hasan, Dr. Pogue and Dr. Celli wish to acknowledge support from the National Cancer Institute (NIH/NCI) P01CA084203-06 (T.H., B.W.P.) 1RC1CA146337 (T.H.), R01CA158415 (T.H.), R01CA1609998 (T.H.), K99CA155045 (J.P.C.) and R00CA155045 (J.P.C.).
Author information
Authors and Affiliations
Contributions
J.P.C., I.R., B.W.P. and T.H. designed research; J.P.C., I.R., A.R.B., I.M. and M.D.G. conducted experiments; J.P.C., I.R., A.R.B. and M.D.G. contributed new analytic tools; J.P.C., I.R. and T.H. wrote the main manuscript text and prepared figures; M.D.G. also created portions of Figure 1; all authors reviewed and revised the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Figures
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Celli, J., Rizvi, I., Blanden, A. et al. An imaging-based platform for high-content, quantitative evaluation of therapeutic response in 3D tumour models. Sci Rep 4, 3751 (2014). https://doi.org/10.1038/srep03751
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep03751
This article is cited by
-
Automated spheroid generation, drug application and efficacy screening using a deep learning classification: a feasibility study
Scientific Reports (2020)
-
Exploiting the potential of commercial digital holographic microscopy by combining it with 3D matrix cell culture assays
Scientific Reports (2020)
-
3D microgroove electrical impedance sensing to examine 3D cell cultures for antineoplastic drug assessment
Microsystems & Nanoengineering (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
