
Abstract
Due to its constitutive activity and ubiquitous distribution, CK2 is the most pleiotropic kinase among the individual members of the protein kinase superfamily. Identification of CK2 substrates is vital to decipher its role in biological processes. However, only a limited number of CK2 substrates were identified so far. In this study, we developed an integrated phosphoproteomics workflow to identify the CK2 substrates in large scale. First, in vitro kinase reactions with immobilized proteomes were combined with quantitative phosphoproteomics to identify in vitro CK2 phosphorylation sites, which leaded to identification of 988 sites from 581 protein substrates. To reduce false positives, we proposed an approach by comparing these in vitro sites with the public databases that collect in vivo phosphorylation sites. After the removal of the sites that were excluded in the databases, 605 high confident CK2 sites corresponding to 356 proteins were retained. The CK2 substrates identified in this study were based on the discovery mode, in which an unbiased overview of CK2 substrates was provided. Our result revealed that CK2 substrates were significantly enriched in the spliceosomal proteins, indicating CK2 might regulate the functions of spliceosome.
Similar content being viewed by others

Introduction
CK2 (casein kinase II) is one of the most pleiotropic serine/threonine protein kinases. It is involved in transcription, signaling, proliferation and in various steps of cell development1. Its abnormally elevated levels are correlated to most tumors2. Unlike other protein kinases, CK2 is constitutively active and ubiquitously distributed in eukaryotes and thus its protein substrates make up a substantial proportion of the phosphoproteome3,4. Identification of CK2 substrates is vital to decipher its role in biological processes, especially in diverse diseases including cancer2. However, only a few hundred substrates were identified so far, which was believed to be just the tip of an iceberg5. Mass spectrometry (MS) is a powerful tool to identify proteins and localize phosphorylation sites. It has played a vital role in identifying kinase substrates. In the early studies, the detection of protein phosphorylation relied on introducing radioisotopes into substrate during kinase assay and MS was only used to identify the proteins6,7,8,9,10. Recently, high throughput proteomics techniques were applied to screen putative kinase substrates11,12,13,14. For example, by combining quantitative phosphoproteomics and in vitro kinase reaction in solution, Huang et al.13 identified 61 and 12 potential substrates for PKA and PKG respectively. Compared with using of radio-isotopes to detect in vitro phosphorylation events, quantitative phosphoproteomics has the advantages of safety and high throughput. Because some additives such as ATP and buffer solution were not compatible with downstream sample preparation and MS analysis, the sample preparation steps, which are time-consuming, were required. Recently, we developed a solid phase kinase reaction to screen in vitro kinase substrates15. The proteins in the cell lysate were immobilized onto the agarose beads and used as the protein library to screen kinase substrates. This solid phase approach facilitated the buffer exchange and avoided the cumbersome sample purification steps. However, the substrates identified by all of above in vitro methods have high rate of false positives. To reduce this rate, in vivo evidences are required. A huge number of in vivo phosphorylation events have been detected by large scale phosphoproteomics analysis in different cell types and tissues16,17,18,19,20. These phosphoproteome events can be employed to complement the substrate screening.
In this study, the isotope dimethyl labeling based quantitative phosphoproteomics was combined with the kinase reaction with immobilized proteomes to screen the CK2 kinase substrates in vitro. Specifically, the proteins from cell lysate were immobilized on the sepharose beads and then used as the protein library for the kinase reaction. Compared with control experiment, the phosphorylation sites (p-sites) with significantly higher intensity (up-regulated) were considered as the potential targets of CK2. To remove the sites that may not happen in vivo, these in vitro sites were further searched in the dataset of in vivo p-sites identified by large-scale phosphoproteomics and the sites found in the dataset were retained. By applying this refining procedure, totally 605 high confident CK2 sites corresponding to 356 proteins were obtained.
Results
Integrated workflow for the screening of CK2 substrates
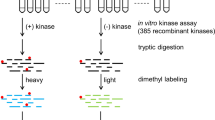
As shown in Figure 1, the integrated workflow has two major steps. The first step is to identify in-vitro kinase substrates. Kinase reaction is performed by using an immobilized proteome as the protein library followed by the identification of putative substrates and their phosphorylation sites using quantitative phosphoproteomics. Detailedly, all the proteins in the cell lysate are coupled onto sepharose beads followed with dephosphorylation of endogenous phosphorylation by the alkaline phosphatase. Next, the dephosphorylated proteins on sepharose beads are divided into two aliquots. One aliquot is used for in vitro kinase reaction by incubating with ATP and CK2 kinase and another aliquot is used for control reaction by incubating with all the same reagents but the CK2 kinase. After the reactions, the peptides are released from the beads by on-bead digestion with trypsin. The peptides derived from the control and kinase reaction are labeled with light (L) and heavy (H) dimethyl labels, respectively. After combining the labeled peptides, the phosphopeptides in the mixture were enriched by Ti4+-IMAC and quantified by online 2D LC-MS/MS. The p-sites specifically generated by CK2 are finally distinguished through the Ratio (H/L) of the quantified sites. This approach will result in the identification of huge number of in vitro CK2 p-sites from hundreds of putative substrates. To improve the identification confidence for substrate screening, a crucial second step is then applied to filter above in vitro dataset. In this step, the in vitro CK2 p-sites are compared with the in vivo p-site dataset acquired by a variety of phosphoproteomics studies in literatures and only the overlapped sites are kept. Because these p-sites are detected in vivo and are the products of CK2 in vitro, they are probable CK2 sites in vivo. By applying this workflow, several hundreds of CK2 substrates and p-sites were identified in this study.

The integrated phosphoproteomics workflow developed for global screening of CK2 kinase substrates.
Large scale identification of in vitro CK2 kinase substrates
We investigated the performance of the first step of the integrated workflow, i.e. the identification of in vitro CK2 kinase substrates. The immobilized proteome was prepared by using proteins (2 mg) in Hela cell lysate. Two parallel experiments were performed. The resulted raw data files were searched and quantified with MaxQuant software21. Totally, 872 and 715 p-sites were quantified from experiments 1 and 2, respectively. Their log2 (Ratio H/L) distributions are shown in Figure 2a. Compared with the control reaction, a large proportion of p-sites were up-regulated after the CK2 reaction. For both experiments, about 30% of the p-sites were quantified with Ratio (H/L) > 2.0, while only about 1% of the p-sites with Ratio (H/L) < 0.5 (see Supplementary Fig. S1 online). These results demonstrated that a significant fraction of these p-sites were in vitro generated by CK2.

(A) Distribution of the log2 (Ratio H/L) of the p-sites quantified in the in vitro experiments 1 and 2. The x-axis indicates the number of p-site identifications and the y-axis represents the log2 (Ratio H/L) of the p-sites. (B) Sequence logos of p-sites quantified with different Ratio (H/L) from experiment 1, the height of each amino acid reflects its occurrence frequency at the corresponding position.
The up-regulated sites are more likely to be CK2 sites. A ratio threshold should be determined to filter the data and distinguish the sites specifically generated by CK2. The threshold was determined based on the fact/observation that CK2 kinase preferentially phosphorylates the sequences with acidic motif5. Accordingly, the quantified p-sites were classified into three groups based on their Ratio (H/L). As shown in Figure 2b, for the p-sites with Ratio (H/L) less than 1.5, the distribution of the amino acid residues around the p-sites was very random except the high frequency of proline at −1 to +3 positions, especially over 50% of proline at +1 position. This profile was similar to that generated by the large-scale phosphoproteome, indicating that these sites were the residual endogenous sites that were not dephosphorylated by alkaline phosphatase. While the sequence logo was significantly different for the sites with Ratio (H/L) between 1.5 and 2.0, D and E mostly occupied the positions at +1 and +3. This profile was similar to the CK2 specificity profile. It should be noted that there was still a relative high frequency of proline present at +1 position, indicating a significant fraction of these sites were not generated by CK2. However, for the sites with Ratio (H/L) higher than 2.0, all the positions were predominated by D/E. Especially, the D/E showed a frequency higher than that of all the other residues together at the +1 and +3 positions. This profile agreed well with that of CK2 reported by Meggio et al.5. Based on the above comparisons, using the threshold of Ratio (H/L) as 2.0 should be able to identify the sites specifically generated by CK2 during the kinase reaction.
The approach was then applied to the large scale analysis of in vitro substrates and p-sites for CK2. The proteins from two cell lysates, i.e. HeLa cell and Jurkat cell, were immobilized onto sepharose beads respectively and three 2D LC-MS/MS replicate runs were performed for analysis of samples from each cell lines. The results were combined and the average Ratio (H/L) were calculated for all the p-sites (see Supplementary Table S1 online). For the p-sites that were identified more than twice in the triplicate MS runs, the relative standard deviation (RSD) of the Ratio (H/L) was calculated. As shown in Supplementary Figure S2 online, more than 90% of these p-sites were quantified with RSD < 50%, this demonstrated the high quantification accuracy of our approach. To generate the high quality in vitro CK2 substrate site dataset, the following criteria were adopted. Firstly, only significantly up-regulated sites (Ratio (H/L) > 2.0) were kept. Secondly, for the p-sites quantified more than once, the sites with RSD > 50% were discarded, unless the Ratio (H/L) of the sites had the same change tendency. Third, the p-sites with localization probability less than 0.5 were discarded.
We further assessed the quality of the in vitro CK2 sites dataset by analyzing of the percentages of acidic residues (D/E/X) around the p-site. Generally, the sequences centered with the p-site (13 residues) were extracted and the phosphorylated sites (pS, pT and pY) other than the central site were replaced as X in the multiple phosphorylated peptides. As shown in Figure 3A, the percentages of acidic residues (D/E/X) on each position of the 13-residue peptide sequences were determined. Similar to the previous study5, the frequency of acidic residue at the +1 and +3 positions were higher than 70%. The numbers of acidic residues at positions from −1 to +5 for these sequences were summarized. It was found that 90.9% of p-sites were surrounded with at least two acidic residues. This further indicated that CK2 preferably phosphorylated the sites with a cluster of acidic residues. However, it should be noted that there were still some p-sites (9.1%) that contained no or one acidic residue at positions from −1 to +5, which meant that these p-sites did not conform to classic CK2 substrate specificity. Therefore, to make the dataset much more confident, we further adapted two criteria to filter the dataset. Firstly, there should be at least two acidic residues at the positions from −1 to +5; and secondly there should be at least one acidic residue at the +1 or +3 position. By this way, totally 988 p-sites corresponding to 581 proteins were screened as CK2 in vitro sites and substrates, respectively (see Supplementary Table S2 online).

(A) Frequency of acidic residues (DEX) in the sequence around the CK2 p-sites. (B) MALDI-TOF MS spectra of the peptide EEQGEGSEDEWEQ incubated with ATP. (C) MALDI-TOF MS spectra of the peptide EEQGEGSEDEWEQ incubated with ATP and CK2. (D) Venn diagram indicates the overlap between the known CK2 substrates dataset and the Class H dataset.
We then investigated if the peptide sequences centered with these determined p-sites could be phosphorylated by CK2 in vitro at peptide level. We randomly selected 12 peptides 1(Table 2). Among these peptides, 9 peptides were phosphorylated by CK2 at the expected sites as detected by the MALDI-TOF MS spectra (see Supplementary Figure S3–S10 online). Take the peptide EEQGEGSEDEWEQ as an example. As shown in Figure 3B and 3C, after the peptide was incubated with CK2, there was a mass shift of 80 Da, indicating phosphorylation of the peptide by CK2. Majority of these peptides could be phosphorylated by CK2, this indicated that the in vitro CK2 kinase substrates determined in this study were of high confidence. However, the phosphorylation of other three peptides was not detected, which was inconsistent to phosphorylation of these sites by CK2 at protein level (all with more than 5-fold increase). This discrepancy for phosphorylation of these sites by CK2 at protein and peptide level may be related with the tertiary structure of protein. For some proteins, the unique protein structure may enable the accumulation of CK2 to the site and the high local concentration of CK2 leads to phosphorylation of the sites on the proteins, even the corresponding sites cannot be phosphorylated by CK2 at peptide level.
Generation of high confident CK2 substrate sites
The phosphorylation on the sites generated by in vitro kinase reaction may not happen in vivo, because the proteins and the kinase may have no chance to touch each other in vivo. Due to the lack of biological context, such as cellular co-localization and/or co-expression of kinases and their substrates, many p-sites identified by in vitro kinase assay are false positives. To improve the confidence of substrate identifications, it is critical to remove false positives. The bona fide CK2 substrate sites must be phosphorylated in vivo in some cell types under certain states and these sites could be identified by comprehensive phosphoproteomics approaches. Up to now, tens of thousands of in vivo p-sites have been identified and collected into three online databases including PHOSIDA22, Phospho. ELM23 and PhosphoSitePlus16. Considering the comprehensiveness of these databases, if the CK2 substrate sites identified above by the in vitro kinase reaction are not present in the databases, these sites are most likely false positives. According to this reasoning, we purified the in vitro results by comparing with the in vivo p-sites of the databases. As shown in Table 1, about 38.8% (383) of the in vitro sites were not included in these databases. These sites were of low confidence to be CK2 in vivo substrate sites and were termed as Class L dataset. After removal of these sites, 605 high confident CK2 substrate sites corresponding to 356 proteins were obtained and these sites and substrates were termed as Class H dataset (see Supplementary Table S2 online).
Discussion
Current large scale phosphoproteomics approach allows identification of numerous cellular phosphoproteins and their p-sites. With the explosion of in vivo phosphorylation events detected, one of the most immediate challenges is delineating of the p-sites to their effector kinases. Motif analysis of these sites showed a high proportion of sites phosphorylated by acidic kinases, such as CK2. However, efforts to validate these sites as CK2 direct substrate sites are hindered by the lack of high throughput in vitro screening methods. In this study, we developed a solid phase based in-vitro approach to screen CK2 substrates in a high throughput way. The use of quantitative phosphoproteomics enabled the identification of hundreds of in-vitro CK2 substrate sites. For the high frequency of phosphorylation events by CK2 in a biological system, when the in vivo sites identified by phosphoproteomics approaches overlapped with the in-vitro sites, these overlapped sites are most likely to be phosphorylated in vivo by CK2.
Comparison with the known CK2 substrates
In this study, many high confident CK2 substrates were identified, it is of interest to compare them with the known CK2 substrates. Therefore, we collected the known CK2 substrates from literatures and totally 328 known CK2 substrates with 705 known p-sites were obtained (see Supplementary Table S3 online). In addition, there were over 100 known CK2 substrates without site information, which were mainly collected in a review by Meggio et al.5. Finally, 58 known CK2 sites corresponding to 39 CK2 substrates were found in our dataset (see Supplementary Table S4 online). As shown in Figure 3D, 36 known CK2 substrates (92%) were from the Class H dataset, indicating the high confidence of CK2 substrates in the Class H dataset. In addition, another 29 known CK2 substrates without identification of the known CK2 p-sites were found and mostly were from the Class H dataset (see Supplementary Table S5 online). This may be caused by the two reasons. First, for some of the 29 proteins, such as protein SSRP1, SIRT1, ABC50 and WASP, the sites identified in our study were not the known CK2 p-sites reported in the literatures. We supposed that there may be multiple CK2 sites for a single protein. Take protein SIRT1 as an example, Ser659 and Ser661 at the C-termini were identified as the p-sites of CK2 kinase with directed site mutations24. Due to the high molecular weight of tryptic peptide containing Ser659 and Ser661, both sites were not identified in our study. However, Thr719 locating on a much smaller tryptic peptide was identified as CK2 p-sites on SIRT1 in our study. The second reason was that, some of the 29 proteins were identified as CK2 known substrates without site information in the literatures. For example, the nuclear protein HIRIP3 had been found to co-purify with CK2 activity and efficiently phosphorylated by CK2 in vitro, however no accurate phosphorylation sites were identified so far25. In this study, 14 CK2 p-sites were identified on HIRIP3. In 2008, Meng et al.26 used protein array to identify the CK2 kinase substrates and they identified protein RDBP, PDCD4 and DDX54 as CK2 substrates, however no site information was obtained. These proteins were all identified as CK2 substrates in our study with accurate site information. Totally, 68 known CK2 substrates were identified in this study and most of the proteins (86.8%) were from the Class H dataset. Above results demonstrated that the CK2 substrates in the Class H dataset had a high probability to be CK2 bona fide substrates.
It is well known that screening of the kinase substrates by conventional approach is time consuming and lab intensive and only a few substrates can be identified in an experiment. This may be the main reason that only 705 CK2 substrate sites were identified cumulatively in literatures, while 605 high confident CK2 substrate sites were identified in this study. This indicated the high throughput of this newly developed approach. However, it should be noted that the overlap between the high confident substrates in Class H dataset and the known CK2 substrates was about 11% (Figure 3D). We supposed that three reasons may be related to the low overlap. Firstly, the known CK2 substrates were identified from different cell lines and different species, while only two human cell lines were used in this study. Some substrates may be not expressed in the two cell lines, or not conserved among different organisms27. Secondly, the known substrates are only a tiny fraction of the all CK2 substrates in biological systems. Thirdly, the immobilization of proteins may make some sites inaccessible to CK2 kinase due to the steric hindrance.
CK2 substrates interact with CK2
As the bona fide substrate, it must be able to touch the kinase in some states. Therefore, the proteins which can interact with CK2 in vivo displayed much higher possibility to be CK2 substrates. Some proteins identified in this study were found associated with CK2 kinase in vivo. In 2002, Gaven et al.28 performed a large scale characterization of the multiprotein complexes in Saccharomyces cerevisiae using tandem-affinity purification and mass spectrometry. They defined 232 distinct multiprotein complexes and the subunits of CK2 kinase appeared in many of these protein complexes. For example, RTF1, LEO1, CTR9 and PAF1 were found in a protein complex containing CK2. All the four proteins were identified as CK2 substrates in our study. Especially, the protein LEO1 was identified with 13 CK2 p-sites. YPR133C (alternative name IWS1) was also found in an immune complex containing CK2 and Spt and it was identified with 13 CK2 p-sites in this study. In 2013, Markku et al.29 performed a rigorous inter-laboratory comparative analysis of the interactomes of 32 human kinases by a standardized AP-MS workflow. They identified 60 proteins that associated with CSNK2A2 (CK2) with high screening criteria. 15 of these proteins (25%) were identified as CK2 substrates in this study, including 2 known CK2 substrate proteins, i.e. DEK and HIRIP3. This clearly indicated that that the other 13 proteins showed high confidence to be CK2 substrates. Above results demonstrated that the CK2 substrates identified in this study had a high probability to be CK2 bona fide substrates.
CK2 targets the splicing machinery
To investigate if CK2 targets any macromolecular complexes, we analyzed the substrates in the Class H dataset by using the Comprehensive Resource of Mammalian Protein Complexes (CORUM), a database of manually curated and validated mammalian protein complexes30. A total of 46 complexes were enriched for the dataset with p-value < 0.05 (Hypergeometric test)31 (see Supplementary Table S6 online). Among them, six had been enriched with a p-value < 0.001. They were spliceosome, C complex spliceosome, CDC5L complex, toposome, LARC complex and MeCP1 complex. It was found that almost all the six complexes were involved in RNA/DNA metabolic process and mRNA splicing. And spliceosome was found to be enriched with a lowest p-value less than 0.0000000002. It is well known that spliceosome is a highly dynamic, macromolecular machine removing noncoding introns from precursor messenger RNAs. In the CPRUM database, 143 proteins were annotated as the components of the spliceosome. It was found a significant fraction (over 20%) of these spliceosomal proteins belong to CK2 substrates. During the course of splicing, an ordered evolution of intermediate splicing complexes designated as complex A (prespliceosome), B (precatalytic spliceosome), B* (activated spliceosome), C (catalytic step 1 spliceosome) and P (post spliceosomal complex) are assembled32. These intermediate splicing complexes vary significantly in their composition. Recently, spliceosome database (see Supplementary Table S7 online) was built by collecting spliceosome-associated proteins identified from a variety of MS experiments33. In order to explore the potential role of CK2 on the dynamic assembly process of the spliceosome, we extracted the proteins in individual spliceosome components from the database and investigated the presence of CK2 potential substrates in the major spliceosomes. As shown in Figure 4, every major spliceosome have more than 8 CK2 substrates. During the course of the splicing reaction, a large number of additional spliceosomal proteins in addition to snRNPs are recruited during the complex transitions. Compared with snRNP subcomplex, 12 of CK2 substrates were observed as the recruited proteins for spliceosome. All these results indicated that CK2 might play an important role in the spliceosome.

The spliceosomes for cross-intron assembly and disassembly cycle, only the major spliceosomal complexes in mammalian splicing extracts are shown.
The number of identified CK2 substrates and the number of proteins in each spliceosome complex were noted as red and blue respectively.
In conclusion, we proposed an integrated phosphoproteomics workflow for global screening of CK2 kinase substrates. Totally 605 high confident CK2 sites corresponding to 356 proteins were identified, the inventory of CK2 substrates will enable better understanding of cellular behaviors that were regulated by CK2. It was found that CK2 substrates were significant enriched in the spliceosome, indicating CK2 might play an important role in the assembling of spliceosome.
Experimental section
Solid phase kinase reaction and on-bead protein digestion
The detailed procedures for the preparation total cell lysate, the immobilization of proteins onto sepharose beads and the dephosphorylation of the immobilized proteins were given in the supplementary materials. The solid phase in vitro kinase reaction was performed similar to that described before15. In brief, the beads with immobilized proteins were suspended in 1 mL kinase buffer (140 mM NaCl, 10 mM MgCl2, 0.1 mM EDTA, 5 mM DTT, 0.1% Triton, 20 mM HEPES (pH 7.6)) at 30°C for 30 min. In vitro kinase reaction was performed by adding of 1 μg casein kinase 2 (Millipore) and 100 μM ATP (Sigma) to the solution. The reaction was allowed to be proceeded for 5 h and then terminated by washing away the kinase buffer by 100 mM ammonium bicarbonate (NH4CO3). After in vitro reaction, the sepharose beads were sequentially incubated with 20 mM DTT and 40 mM IAA in 100 mM TEAB buffer (pH = 8.0). Then trypsin was added and the digestion was performed at 37°C overnight. After digestion, the supernatant was collected. The sepharose were further completely washed with TEAB buffer for three times. Finally, the solutions were combined. The control experiment was performed as above except no CK2 was added.
Dimethyl labeling and phosphopeptides enrichment
For the light and heavy dimethyl labeling, 240 μL of CH2O (4%, v/v) and CD2O (4%, v/v) were added into the digests of control and kinase samples respectively and then 240 μL of freshly prepared NaBH3CN (0.6 M) were added subsequently to both samples. The resultant mixture was incubated for 1 h at room temperature. And then 20 μL of ammonia (25%, v/v) was added to each mixture for 15 min. After that, 10% TFA solution was added to each mixture to adjust the pH to 2 ~ 3 and the two mixtures were mixed for further phosphopeptides enrichment. The phosphopeptides were enriched by Ti4+-IMAC microspheres following the protocol reported by Zhou et al.34.
MS and data analysis
The 2D-RPLC-MS/MS was performed with a series of stepwise elution with salt concentrations of 0, 25, 50, 100, 1000 mM NH4AC. Detailed information was given in the supplementary materials. The raw data files were analyzed with MaxQuant software (version 1.1.1.36)21. The required FDR was set to 0.01 at the peptide, protein and site level, respectively. All the MS/MS spectra for the identified phosphopeptides were exported to be. png files by the MaxQuant software and available in the PeptideAtlas database (ftp://PASS00260:AF642ga@ftp.peptideatlas.org/). The ion chromatograms for a few randomly selected peptides labeled with light (CH3, black line) and heavy (CHD2, red line) dimethyl labels were checked and no obvious retention time shift was observed (Supplementary Figure S11). Sequence logos were generated by the WebLogo program35.
Comparison with the known CK2 substrates
We compared the substrates identified in this study with the known CK2 substrates in the database by the following way. Firstly, we compared the 13-residue sequences around the p-sites identified in this study with the sequences containing CK2 p-sites of the known CK2 substrates in the database. The overlapped proteins were retained. Then, we compared the other information such as “Substrate ACC (UniProt ID)” and “Gene Symbol”, only the proteins with the same description were kept. The positions of p-sites were also compared. However, as the length of some proteins are different in the different versions of databases, the position of the site reported in this study is based on the IPI database. For comparison with known substrates in species other than human, we used the blast tool of the Uniprot database and the online database named PhosphoSitePlus (mostly used)16 to find the orthologs of the protein from other species. For the sites commonly belonged to different protein isoforms, all the protein IDs were listed in the results.
CORUM complex enrichment analysis
The protein complex annotations for human dataset were downloaded from CORUM database30 (http://mips.gsf.de/genre/proj/corum/index.html), which contained manually curated and experimentally verified protein complex annotations. The IPI identifiers were mapped to UniProt identifiers to enable comparison with CORUM database. Hypergeometric test was used to filter enriched complexes with respect to the complete CORUM human dataset, p-value < 0.05 was considered as a significant enrichment. The proteins for intermediate splicing complexes were downloaded from the website (http://spliceosomedb.ucsc.edu)33.
References
Olsten, M. E. K. & Litchfield, D. W. Order or chaos? An evaluation of the regulation of protein kinase CK2. Biochem Cell Biol 82, 681–693 (2004).
Kramerov, A. A. & Ljubimov, A. V. Focus on Molecules: Protein kinase CK2. Exp Eye Res 101, 111–112 (2010).
Litchfield, D. W. Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem J 369, 1 (2003).
Faust, M. & Montenarh, M. Subcellular localization of protein kinase CK2. A key to its function? Cell Tissue Res. 301, 329–340 (2000).
MEGGIO, F. & PINNA, L. One-thousand-and-one substrates of protein kinase CK2? FASEB J 17, 349 (2003).
Allen, J. J., Lazerwith, S. E. & Shokat, K. M. Bio-orthogonal affinity purification of direct kinase substrates. J Am Chem Soc 127, 5288–5289 (2005).
Shah, K., Liu, Y., Deirmengian, C. & Shokat, K. M. Engineering unnatural nucleotide specificity for Rous sarcoma virus tyrosine kinase to uniquely label its direct substrates. Proc Natl Acad Sci U S A 94, 3565–3570 (1997).
Knebel, A., Morrice, N. & Cohen, P. A novel method to identify protein kinase substrates: eEF2 kinase is phosphorylated and inhibited by SAPK4/p38¦Ä. EMBO J 20, (2001).
Li, X. et al. The reverse in-gel kinase assay to profile physiological kinase substrates. Nat Methods 4, 957–962 (2007).
Troiani, S. et al. Searching for biomarkers of Aurora-A kinase activity: identification of in vitro substrates through a modified KESTREL approach. J Proteome Res 4, 1296–1303 (2005).
Wang, P. et al. Quantitative phosphoproteomics identifies SnRK2 protein kinase substrates and reveals the effectors of abscisic acid action. Proc Natl Acad Sci U S A 110, 11205–11210 (2013).
Xue, L. et al. Sensitive kinase assay linked with phosphoproteomics for identifying direct kinase substrates. Proc Natl Acad Sci U S A 109, 5615–5620 (2012).
Huang, S. Y., Tsai, M. L., Chen, G. Y., Wu, C. J. & Chen, S. H. A systematic MS-based approach for identifying in vitro substrates of PKA and PKG in rat uteri. J Proteome Res 6, 2674–2684 (2007).
Knight, J. D. R. et al. A novel whole-cell lysate kinase assay identifies substrates of the p38 MAPK in differentiating myoblasts. Skeletal muscle 2, 5 (2012).
Zhang, M. Y. et al. A bead-based approach for large scale identification of in vitro kinase substrates. Proteomics 11, 4632–4637 (2011).
Hornbeck, P. V. et al. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res 40, D261–270 (2012).
Dephoure, N. et al. A quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci U S A 105, 10762 (2008).
Lundby, A. et al. Quantitative maps of protein phosphorylation sites across 14 different rat organs and tissues. Nat Commun 3, 876 (2012).
Olsen, J. et al. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Science's STKE 3, ra3 (2010).
Huttlin, E. L. et al. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189 (2010).
Cox, J. & Mann, M. MaxQuant enables high peptide identification rates, individualized ppb-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26, 1367–1372 (2008).
Gnad, F., Gunawardena, J. & Mann, M. PHOSIDA 2011: the posttranslational modification database. Nucleic Acids Res 39, D253–D260 (2011).
Dinkel, H. et al. Phospho. ELM: a database of phosphorylation sites–update 2011. Nucleic Acids Res 39, D261–D267 (2011).
Zschoernig, B. & Mahlknecht, U. Carboxy-terminal phosphorylation of SIRT1 by protein kinase CK2. Biochem Biophys Res Commun 381, 372–377 (2009).
Assrir, N., Filhol, O., Galisson, F. & Lipinski, M. HIRIP3 is a nuclear phosphoprotein interacting with and phosphorylated by the serine-threonine kinase CK2. Biol Chem 388, 391–398 (2007).
Meng, L. et al. Protein kinase substrate identification on functional protein arrays. BMC Biotechnol 8, 22 (2008).
Gupta, R. et al. Ubiquitination screen using protein microarrays for comprehensive identification of Rsp5 substrates in yeast. Mol Syst Biol 3, (2007).
Gavin, A. C. et al. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 415, 141–147 (2002).
Varjosalo, M. et al. Interlaboratory reproducibility of large-scale human protein-complex analysis by standardized AP-MS. Nat Methods 10, 307–314 (2013).
Ruepp, A. et al. CORUM: the comprehensive resource of mammalian protein complexes--2009. Nucleic Acids Res 38, D497–501 (2010).
Choudhary, C. et al. Lysine Acetylation Targets Protein Complexes and Co-Regulates Major Cellular Functions. Science 325, 834–840 (2009).
Wahl, M. C., Will, C. L. & Luhrmann, R. The spliceosome: design principles of a dynamic RNP machine. Cell 136, 701–718 (2009).
Cvitkovic, I. & Jurica, M. S. Spliceosome Database: a tool for tracking components of the spliceosome. Nucleic Acids Res 41, D132–D141 (2013).
Zhou, H. J. et al. Specific phosphopeptide enrichment with immobilized titanium ion affinity chromatography adsorbent for phosphoproteome analysis. J Proteome Res 7, 3957–3967 (2008).
Crooks, G. E., Hon, G., Chandonia, J. M. & Brenner, S. E. WebLogo: a sequence logo generator. Genome Res 14, 1188 (2004).
Acknowledgements
We thank Dr. Lei Li for proof reading, Manyu Zhang for helpful discussions and Jing Dong for synthesis of Ti4+-IMAC microspheres.
Author information
Authors and Affiliations
Contributions
H.F.Z. and M.L.Y. designed experiments; Y.Y.B. performed most of the experiments; C.L.W. helped to perform the experiments; Y.Y.B., M.L.Y. and K.C. analyzed data; C.X.S. and M.M.D. participated in quantitative LC-MS/MS analysis; Y.B.P. and Q.H.Q. participated in the 5800 MALDI-TOF/TOF analysis; Y.Y.B., M.L.Y. and H.F.Z. wrote the manuscript. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Funding: This work was supported by the China State Key Basic Research Program Grant (2013CB911202, 2012CB910101, 2012CB910604), the Creative Research Group Project of NSFC (21021004), theNational Natural Science Foundation of China (21275142,21235006, 81161120540), National Key Special Program on Infection diseases (2012ZX10002009-011), Analytical Method Innovation Program of MOST (2012IM030900) to H.F. Zou.
Electronic supplementary material
Supplementary Information
Supplementary Table S1
Supplementary Information
Supplementary Table S2
Supplementary Information
Supplementary Table S3
Supplementary Information
Supplementary Table S6
Supplementary Information
Supplementary Table S7
Supplementary Information
Supporting Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Bian, Y., Ye, M., Wang, C. et al. Global Screening of CK2 Kinase Substrates by an Integrated Phosphoproteomics Workflow. Sci Rep 3, 3460 (2013). https://doi.org/10.1038/srep03460
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep03460
This article is cited by
-
Nutritional stress-induced regulation of microtubule organization and mRNP transport by HDAC1 controlled α-tubulin acetylation
Communications Biology (2023)
-
Molecular targets and therapeutic strategies for triple-negative breast cancer
Molecular Biology Reports (2023)
-
Protein kinase CK2 – diverse roles in cancer cell biology and therapeutic promise
Molecular and Cellular Biochemistry (2023)
-
Phosphoproteomics revealed cellular signals immediately responding to disruption of cancer amino acid homeostasis induced by inhibition of l-type amino acid transporter 1
Cancer & Metabolism (2022)
-
BRCA1 mutations in high-grade serous ovarian cancer are associated with proteomic changes in DNA repair, splicing, transcription regulation and signaling
Scientific Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
