
Abstract
Recent reports on high-mobility organic field-effect transistors (FETs) based on donor-acceptor semiconducting co-polymers have indicated an apparently strong deviation from the paradigm, valid for a series of semi-crystalline polymers, which has been strictly correlating charges mobility to crystalline order. This poses a severe limit on the control of mobility and a fundamental question on the critical length scale which is dominating charge transport. Here we focus on a well-known model material for electron transport, a naphthalene-diimide based copolymer and we demonstrate that mobility can be controlled over two orders of magnitude, with maximum saturation mobility exceeding 1 cm2/Vs at high gate voltages, by controlling the extent of orientational domains through a deposition process as simple as spin-coating. High mobility values can be achieved by adopting solvents inducing a higher amount of pre-aggregates in the solution, which through the interaction with the substrate, provide the polymer with liquid-crystalline like ordering properties.
Similar content being viewed by others
Introduction
Solution-processed, organic semiconductors based field-effect transistors (FETs)1, which may enable portable, lightweight and conformable electronics, are experiencing a boost in performances mainly thanks to the recent development of high-mobility donor-acceptor semiconducting copolymers2,3. Yet, a clear picture of the mechanisms governing charge transport in thin polymeric films, which would enable the control of field-effect mobility (μ), is missing4,5. While in general the degree of crystalline order is recognized to play an important role in sizing field-effect mobility in solution-processed π-conjugated semiconductors6,7, polymer thin films, usually deposited by spin-coating or printing techniques, are inherently prone to disorder. Charge carriers are usually highly localized and denote polaronic features8; importantly they have to drift/percolate in the accumulated channel of a FET for distances from a few to 100 μm in most of devices presented in the literature. In this framework, μ is evidently a figure of merit which averages local properties (molecular structure, packing, electronic couplings), developing at the (sub-)nanometer scale, over distances orders of magnitude larger, thus urging to investigate and control different length scales, from the molecular one up to the macro-scale which characterizes prototypal devices. Very recently the importance of para-crystallinity of the ordered phase within disordered films has been proposed in support of a unifying picture where trapping caused by lattice disorder dictates the charge transport properties in high-mobility polymer semiconductors9. Moreover, long-range orientational order, revealed by soft X-rays techniques, has been reported to nicely correlate with μ for a p-type polymer with a liquid-crystalline meso-phase10. On a larger scale, it was shown that polymers specifically designed to show lyotropic liquid-crystalline behavior can achieve alignment of the conjugated core over large areas, enabling efficient charge transport preferentially along the polymer chain direction11. These studies indicate that the complex microstructure of polymer films influences charge transport at different length-scales.
Recently developed donor-acceptor copolymers12,13, displaying high μ values (>1 cm2/Vs) even in presence of a substantial degree of disorder, excite particular interest. Among these materials, poly{[N,N′-bis(2-octyldodecyl)-naphthalene-1,4,5,8-bis(dicarboximide)-2,6-diyl]-alt-5,5′-(2,2′-bithiophene)} (Figure 1a, P(NDI2OD-T2)) represents a notable example14: it stands for a class of materials which can yield high electron mobility15,16,17 and it is one of the most studied n-channel polymeric semiconductors in the recent literature, displaying electrons mobility from ~0.10 up to 0.96 cm2V−1s−1 14,18. Importantly, despite the several reports on the subject, the control the mobility in the reported range has not been rationalized yet.

Cross-polarized optical microscope image.
Optical microscope image taken through two crossed-polarizers to highlight birefringence in a P(NDI2OD-T2) film deposited from toluene. The areas which appear dark highlights the polymer alignment along one of two polarizes direction, while the narrow bright line identify a direction close to ±45° (maximum transmission light intensity). At the gentle intersection of two or more lines, the polymer order is rearranged resulting in an extended defect region. In the inset: molecular structure of P(NDI2OD-T2).
While most of the studies on P(NDI2OD-T2) devices concentrated on the semiconductor-dielectric interface18,19,20, surprisingly no direct correlation of transport properties with the specific semiconductor microstructure has been reported. Films spun from dichlorobenzene are characterized by the co-presence of an amorphous-like phase and a highly interconnected ordered phase21,22, where polymer backbones lay preferentially flat on the substrate23. For spun films, not treated beyond their melting point, the ordered phase show crystalline planes parallel to the substrate24 (the so called “face-on” structure), in which the conjugated segments lay with a dihedral angle between the NDI and thiophene units23. Melt-annealing treatments allow to drastically modify the orientation of the ordered phase25; however, frustrating common arguments regarding charge transport, mobility values are minimally affected26.
As well as thermal annealing, a common strategy to access and improve molecular self-assembling consists in the accurate choice of the processing solvent and of the deposition/treatment technique employed27,28. Here we demonstrate that in P(NDI2OD-T2) electrons mobility can be controlled over two orders of magnitude, with maximum saturation mobility exceeding 1 cm2/Vs at high gate voltages, with a simple spin-coating process by adopting solvents which introduce a different amount of chains pre-aggregation in the solution. Pre-aggregation refers to the formation of molecular aggregates already in solution, as an effect of the particular solvent adopted, prior to the deposition of the film. Solvents yielding a higher degree of pre-aggregation enable liquid-crystalline like long-range order with exceptionally extended orientational domains, spanning hundreds of micrometers, thus strongly enhancing charges mobility in FETs. By linking transport properties to length scales from the meso- to macro-scale, this work offers a critical perspective for controlling and further improving mobility in polymer FETs.
Results
Formation of P(NDI2OD-T2) birefringent films
A clear effect of the solvent on P(NDI2OD-T2) absorption in solution was observed by Steyrleuthner et al. and imputed to intra-chain aggregation phenomena induced by different solvation properties; however, no relevant differences were observed in the optical absorption of thin films obtained from solutions with variable amount of pre-formed aggregates29. Since UV-Vis optical spectra reflect on average the local molecular conformation and inter-chain interactions of the conjugated segments, the previous observations do not preclude a different morphology of the films on a larger scale length, which we have investigated here.
The different degree of pre-aggregation may instead have a critical role in the large-scale morphology of the deposited films - and then in charge mobility - as plainly evidenced by the polarized light microscope analysis (PLM), a simple contrast-enhancing technique that improves the quality of images obtained with birefringent materials. We have employed as solvents toluene, which induces the highest level of pre-aggregation in solution, 1,2-dichlorobenzene (DCB), where a smaller content of aggregates is present and a mixture of chloronaphthalene and chloroform (CN:CF, 80:20 in volume), which hampers the formation of aggregates29 (Supporting Information, SI, Figure S1). A striking difference emerges in PLM images of the 40–50 nm thick corresponding films deposited by spin-coating: while substantially no features appear in films deposited by CN:CF and DCB (Figure S2), birefringent domains extending even for millimiters have been observed in films deposited by toluene (Figure 1). These domains are optical fingerprints of an order which extends over the macro-scale and their formation suggests a liquid-crystalline order parameter for the polymer chains, which preserve a preferred directionality, i.e. an average direction of the backbone maintained over a certain spatial range, depending on the deposition conditions, such as the spin-speed. While we always observe birefringence in P(NDI2OD-T2) films deposited from aggregates-rich solutions, the elongation of domains can be hampered by too fast spin speeds or by a short interaction time of the aggregates with the substrate (Figure S3).
We verified that the mechanism locking this peculiar morphology is primarily dictated by the amount of aggregates rather than by the solvent boiling point: if we adopt mesitylene as a further solvent, characterized by a much higher boiling point (~164°C) with respect to toluene (~110°C) and closer to DCB one (~180°C), solutions display aggregation features as in toluene and produce birefringent films similar to Figure 1 when deposited by spin-coating at similar spin-speeds (Figure S4). While ordering of functional polymer films is generally achieved with specific alignment techniques30, it is rather remarkable that such morphology can be obtained already by means of spin-coating, on bare glass substrates without particular surface or thermal treatments. Indeed, we observe the formation of the birefringent pattern immediately after spinning at room temperature for just 30 seconds, where a relevant amount of solvent is still present in the film. Drop-casting indicate that the pattern forms at the interface with the substrate already when a substantial amount of the solutions is still drying (Figure S4e), confirming the strong effect of aggregates-substrate interaction in defining the film morphology.
Orientational order as the origin of birefringence
We have further analyzed the morphological differences on a smaller scale by means of detailed AFM measurements. The topography of films from CN:CF consists of small nodules, just a few tenths of nanometers large, composing a surface with a Root Mean Square Roughness (RRMS) of 1.9 nm (Figure 2a and b). Differently, films from DCB and toluene display a smoother topography (RRMS ~ 0.6 nm), with supra-molecular fibrils which are 10–20 nm wide (Figure 2c, e), clearly composed, at an even smaller scale, by elongated rod-like features, which appear 2–3 nm wide through convolution with the scanning tip (Figure 2d, f). By comparing DCB and toluene processed samples, a clear and decisive difference in the correlation length of the fibrils emerges: in the case of DCB, fibrils appear to be shorter, more branched and entangled and their directional correlation is apparently lost within several hundreds of nanometers (Figure 2c and d), in agreement with recently reported TEM22 and Charge Modulation Microscopy31 results on similar films. Differently, much longer fibrils are obtained from toluene, where a preferential orientation of the fibrils is preserved over several microns (Figure 2e, f and Figure S5). This orientational order of supra-molecular structures justifies the birefringent features observed with PLM.

Films topography.
AFM images of P(NDI2OD-T2) thin films deposited from CN:CF (a,b), DCB (c,d) and toluene (e,f) solutions. Scan size is 2 μm × 2 μm in (a), (c) and (e) and 0.3 μm × 0.3 μm in (b), (d) and (f). Nodular structures are revealed in films deposited from CN:CF, while fibrils appear in the case of DCB and toluene. In the case of toluene, fibrils preserve a preferential alignment, one with respect to the other, over the all scanned area: this is highlighted by 2D Fast Fourier Transform (FFT) images (modules, in the insets, scale bar: 100 μm−1).
To clarify the relationship between PLM observations and topography, we have further analyzed the films from toluene with a single polarizer (Figure 3) and AFM measurements extended over large areas at the edges between different domains (Figure S5). With the former, it is possible to identify in detail the orientation of the absorbing conjugated segments within polymer chains with respect to the impinging polarized light. When turning the polarizer of 90°, the wider dark areas (polymer backbone parallel to the polarized axis) become bright and vice versa, passing through the narrow lines of conjunction (backbone at 45°) between the two preferential orientations. Correspondingly the AFM analysis highlights the presence of domains that are tens of micrometers large, characterized by two specific alignments of the fibrils, one perpendicular to the other. Since in each domain the absorbance is stronger in the direction parallel to the fibrils, this reveals that the polymer backbone have to be preferentially extended in the direction of the fibrils. The transition between the two fibrils directions is not sharp, but is achieved through a smooth change of the angle over a distance of ~10–20 μm (Figure S5): it is within this transition area that at 45° it is possible to observe neat, 5 to 10 μm wide lines through a polarizer which separates the wider domains. Within these marked lines fibrils can keep parallel to each other over samples as wide as 1–2 cm2.

Polarized optical microscope image.
(a) Optical microscope image taken using one polarizer to identify preferential orientation within the ordered domains of P(NDI2OD-T2) film deposited from toluene. Starting from the top, the polarizer is rotated incrementally by 45°. Images at smaller intervals are provided as a video in the supplementary material. (b) A sketch of P(NDI2OD-T2) polymer backbones orientation (white dashed guidelines) across the domains within the inset indicated in panel (a). Supra-molecular fibrils follow the same orientation, as evidenced by AFM analysis (Figure S5).
Effect of long-range order on charges mobility
Spurred by these compelling observations, we have fabricated Top-Gate, Bottom-Contacts FETs (Figure 4a) to directly assess the impact of the reported macro-scale morphologies on the charge transport properties. All devices show excellent n-type characteristic curves (Figure 4b,c and Figure S6) where the highest currents are consistently measured on films deposited from toluene (mesitylene provides similar results, see SI), followed by DCB. The worst performances are recorded for films from CN:CF. We have extracted μ in the linear and in the saturation regime (Figure 4c) for at least 10 devices with a channel length L = 20 μm for each system. In the linear regime (Vd = 5 V) the average mobility corresponds to 0.279 cm2V−1s−1, 0.242 cm2V−1s−1, 0.071 cm2V−1s−1, 0.015 cm2V−1s−1 for devices from toluene, from mesitylene, from DCB and from CN:CF, respectively (standard deviations: 0.109 cm2V−1s−1, 0.080 cm2V−1s−1, 0.025 cm2V−1s−1, 0.005 cm2V−1s−1 respectively). While linear mobility values are weakly dependent on Vg (for Vg > 10 V, Figure 4d), in the saturation regime (Vd = 50 V) this is true only for the CN:CF case. Saturation mobility shows an evident Vg dependence on devices based on films where fibrils are present, an effect which is stronger in the most aligned samples (i.e. from toluene). For the DCB and toluene case, up to Vg = 15–20 V, saturation mobilities are similar to linear mobilities, while for Vg > 20 V the gate voltage dependence leads to much higher saturation values. At Vg = 50 V, the average saturation mobility corresponds to 0.730 cm2V−1s−1, 0.750 cm2V−1s−1, 0.095 cm2V−1s−1 and 0.020 cm2V−1s−1 for devices from toluene, from mesitylene, from DCB and from CN:CF, respectively (standard deviations: 0.370 cm2V−1s−1, 0.495 cm2V−1s−1, 0.050 cm2V−1s−1 and 0.005 cm2V−1s−1 respectively). Within our tests on 20 μm devices, a maximum saturation mobility of 1.5 cm2V−1s−1 was measured on a film deposited from mesitylene at Vg = Vd = 50 V. We have also scaled the channel lengths of devices to further investigate the gate voltage dependence of mobility (Figure 4f and Figure S7), which is likely due to an effect of the lateral field on injection and/or transport32,33. Linear mobilities do not vary with L from 5 to 20 μm, while the gate voltage dependence of saturation mobility is stronger and stronger with shorter channel lengths, clearly pointing to an effect of the large lateral field dropping over the channel from source to pinch-off point. Owing to this, on 10 μm devices, we can extract mobility values exceeding 1 cm2V−1s−1 for Vg = 30 V, both for samples deposited from toluene and mesitylene; this mobility value is reached already at Vg below 20 V for L = 5 μm.

Effect of long-range order on charge transport properties in P(NDI2OD-T2) FETs.
(a) Schematic representation of the Top-Gate, Bottom-Contacts FETs realized in this work; channel width W = 10 mm, channel length L = 20 μm, dielectric specific capacitance Cdie = 4.8 nFcm−2. (b) N-channel transfer characteristics in saturation regime (Vd = 50 V) of the FETs with P(NDI2OD-T2) films as the active layer deposited from toluene (black line), DCB (red line) and CN:CF (green line). (c) N-channel transfer characteristics in linear regime (Vd = 5 V) with films deposited from toluene (black line), DCB (red line) and CN:CF (green line); dashed blue lines indicate the linear fit. (d) Typical linear (dashed lines) and saturation (solid lines) mobility as a function of the gate voltage for FETs with films processed from different solvents. (e) Average linear and saturation mobility as a function of the solvent for a set of at least 10 devices for each data point. (f) Typical linear and saturation mobility as a function of the gate voltage for FETs with films deposited from toluene with L = 20 μm (blue lines), L = 10 μm (orange lines), L = 5 μm (magenta lines).
Discussion
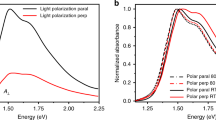
Our data show that charge transport in a high-mobility donor-acceptor copolymer is drastically affected by the film morphology and that the longer the length of the orientational order, the higher is the field-effect mobility. We can suggest that when the long-range order is maintained over distances exceeding the channel length of the devices (5–20 μm), efficient charge transport from source to drain can be observed. One fundamental question regards the nature of the charge carriers and how this is affected by the different film microstructure. To shine light on this aspect, we have performed Charge Modulation Spectroscopy (CMS) measurements34, which probe the charge induced features in the nanometer-thick accumulated channel (Figure 5). The spectra of devices from all employed solvents show similar polaronic absorption features, which have been previously discussed19, with only a slight broadening for toluene and DCB case and no appearance of different transitions. This means that the charge carrier, which is similarly localized in all the cases, is experiencing a comparable local electronic structure. It is therefore possible to conclude that the differences in μ cannot be ascribed to a different nature of the local packing and chain conformation; indeed also UV-vis absorption spectra, here reflected in the positive bleaching band which accounts for transitions of only the ground-state conjugated segments probed by charges, are very similar. Moreover, previous studies reveal that the content of aggregates does not substantially vary from film to film29. This brings us to the conclusion that it is the macro-scale which is sizing the field-effect mobility, underlining the role of long-range order in enabling an efficient percolation pathway for localized polaronic charge carriers through more efficient interconnections of similar aggregates. At the same time, we observe that associated with fibrils within films and their ordering, a gate voltage dependence of mobility develops in the saturation regime. Although further investigations are required, this is an interesting feature, which could indicate that high lateral voltages differently affect the transport properties, depending on the particular morphology of the film and alignment of polymer chains.

Charge Modulation Spectroscopy measurements on P(NDI2OD-T2) FETs.
CMS spectra of films from toluene (red line), DCB (green line) and CN:CF (blue line), measured on semitransparent FETs described in the Methods section.
These results provide important indications for the further improvement of charge transport in polymer FETs in order to meet mobility figures suitable for their wide-spread applications in electronics products. While chemical design of molecular structures yielding higher and higher field-effect mobility is assisting this development, we show how critical parameters, such as pre-aggregation, can be used as powerful tools in order to control and enhance field-effect mobility in thin polymeric films deposited even by a simple spin-coating process, thanks to a solvent-induced, liquid-crystalline like, long-range order.
Methods
Samples and devices preparation
Thoroughly cleaned 1737F glass or SiO2 were used as substrates for all the films realized in this work. FETs were fabricated according to a Top-Gate, Bottom-Contacts architecture. Bottom Au contacts were defined by a lift-off photolithographic process with a 0.7 nm thick Cr adhesion layer. The thickness of the Au contacts was 30 nm. Patterned substrates were cleaned in an ultrasonic bath in isopropyl alcohol for 2–3 min before deposition of the semiconductor or the dielectric. P(NDI2OD-T2) was purchased from Polyera Corporation (Activink™ N2200). Solution of P(NDI2OD-T2) in toluene (5 g/l), mesitylene (5 g/l), DCB (9 g/l) and CN:CF solvent mixture 80:20 (10 g/l in total) were prepared, filtered and deposited by spin-coating at 1000 rpm for 30 s to 90 s in a nitrogen glove box. Only the films from CN:CF were stored overnight in high vacuum (7 × 10−7 mbar) to ensure the complete removal of the solvent. The semiconductor was then annealed for 14 h at 120°C on a hot plate in a nitrogen atmosphere. PMMA (Sigma-Aldrich) with Mw = 120 kg mol−1 was spun from 2-butanone (MEK) (with a concentration of 70 g/l). A dielectric-layer thickness of ~660 nm was obtained. After the dielectric deposition, the devices were annealed under nitrogen, on a hot-plate, at 80°C for 4 h. 30 nm thick gate Al electrodes were thermally evaporated as gate contacts. For CMS measurements, the perfluorinated polymer CYTOP CTL-809M dielectric (Asahi Glass) was spun as received at 6000 rpm for 90 s (film thickness ~ 550 nm) as the dielectric layer and thermally evaporated 4.5 nm thick Au semi-transparent gate electrodes were employed. For UV-Vis absorption of P(NDI2OD-T2) solutions the following concentrations were employed: toluene: 0.5 g/l; mesitylene: 0.05 g/l; DCB: 0.9 g/l; CN:CF: 1 g/l. For PLM and UV-Vis absorption of P(NDI2OD-T2) thin films identical spin coating and annealing procedures as adopted for FETs fabrication were employed.
Films topography and thickness
The surface topography of the films was measured with an Agilent 5500 Atomic Force Microscope operated in the Acoustic Mode. The thicknesses of the polymer films were measured with a KLA Tencor Alpha-Step Surface Profiler.
Electrical characterization
The electrical characteristics of transistors were measured in a nitrogen glovebox on a Wentworth Laboratories probe station with an Agilent B1500A semiconductor device analyzer. Linear charge carrier mobility values were extracted by the transfer characteristic curves according to the gradual channel approximation35, following the expression  , where Id is the drain current, μlin is the linear mobility, Cdie is the specific dielectric capacitance, W and L are the width and the length of the channel, respectively, Vg is the gate voltage, Vd is the drain voltage and Vt is the threshold voltage. Saturation charge carrier mobility values were extracted by the transfer characteristic curves according to the expression
, where Id is the drain current, μlin is the linear mobility, Cdie is the specific dielectric capacitance, W and L are the width and the length of the channel, respectively, Vg is the gate voltage, Vd is the drain voltage and Vt is the threshold voltage. Saturation charge carrier mobility values were extracted by the transfer characteristic curves according to the expression  , where Id is the drain current, μsat is the saturation mobility. Accordingly, the Vg dependent values of μlin (μsat) were obtained from the slope of Id vs. Vg (Id0.5 vs. Vg), calculated every three points around each Vg value.
, where Id is the drain current, μsat is the saturation mobility. Accordingly, the Vg dependent values of μlin (μsat) were obtained from the slope of Id vs. Vg (Id0.5 vs. Vg), calculated every three points around each Vg value.
Optical characterization
Polarized optical microscope images were recorded in transmission mode with a Zeiss Axio Scope A1 equipped with two crossed polarizers (Figure 1) or with a single polarizer through a long-wavelength pass edge filter with cut-off at 600 nm (Figure 3).
Charge modulation spectroscopy
The CMS spectra were collected by measuring the normalized transmittance variation (ΔT/T) induced by applying a modulated voltage (Vg = 60 V ± 10 V, fmod = 993 Hz) to the gate electrode while keeping the source and drain contacts grounded. A tungsten lamp was used as a probing light. Probe light, after being monochromated, was focused on the sample which was kept in a vacuum chamber during the whole measurement. The transmitted light was then collected and focused on a silicon photodiode whose output was connected to a transimpedance amplifier (Femto DHPCA-100); the signal was then recovered through a digital lock-in amplifier (Stanford Research Systems SR830).
References
Klauk, H. Organic thin-film transistors. Chem. Soc. Rev. 39, 2643–2666 (2010).
Baeg, K.-J., Caironi, M. & Noh, Y.-Y. Toward Printed Integrated Circuits based on Unipolar or Ambipolar Polymer Semiconductors. Adv. Mater. 25, 4210–4244 (2013).
Nielsen, C. B., Turbiez, M. & McCulloch, I. Recent Advances in the Development of Semiconducting DPP-Containing Polymers for Transistor Applications. Adv. Mater. 25, 1859–1880 (2013).
Asadi, K. et al. Polaron hopping mediated by nuclear tunnelling in semiconducting polymers at high carrier density. Nat. Commun. 4, 1710 (2013).
Mehraeen, S., Coropceanu, V. & Brédas, J.-L. Role of band states and trap states in the electrical properties of organic semiconductors: Hopping versus mobility edge model. Phys.l Rev. B 87, 195209 (2013).
Diao, Y. et al. Solution coating of large-area organic semiconductor thin films with aligned single-crystalline domains. Nat. Mater. 12, 665–671 (2013).
McCulloch, I. et al. Liquid-crystalline semiconducting polymers with high charge-carrier mobility. Nat. Mater. 5, 328–333 (2006).
Sirringhaus, H., Bird, M. & Zhao, N. Charge transport physics of conjugated polymer field-effect transistors. Adv. Mater. 22, 3893–3898 (2010).
Noriega, R. et al. A general relationship between disorder, aggregation and charge transport in conjugated polymers. Nat. Mater. 12, 1038–1044 (2013).
Collins, B. A. et al. Polarized X-ray scattering reveals non-crystalline orientational ordering in organic films. Nat. Mater. 11, 536–543 (2012).
Kim, B.-G. et al. A molecular design principle of lyotropic liquid-crystalline conjugated polymers with directed alignment capability for plastic electronics. Nat. Mater. 12, 659–664 (2013).
Lee, J. et al. Boosting the Ambipolar Performance of Solution-Processable Polymer Semiconductors via Hybrid Side-Chain Engineering. J. Am. Chem. Soc. 135, 9540–9547 (2013).
Li, J. et al. A stable solution-processed polymer semiconductor with record high-mobility for printed transistors. Sci. Rep. 2 (2012).
Yan, H. et al. A high-mobility electron-transporting polymer for printed transistors. Nature 457, 679–686 (2009).
Guo, X., Kim, F. S., Seger, M. J., Jenekhe, S. A. & Watson, M. D. Naphthalene Diimide-Based Polymer Semiconductors: Synthesis, Structure–Property Correlations and n-Channel and Ambipolar Field-Effect Transistors. Chem. Mater. 24, 1434–1442 (2012).
Kim, R. et al. High-Mobility Air-Stable Naphthalene Diimide-Based Copolymer Containing Extended π-Conjugation for n-Channel Organic Field Effect Transistors. Adv. Funct. Mater., in press; DOI: 10.1002/adfm.201301197 (2013).
Luzio, A. et al. Synthesis, Electronic Structure and Charge Transport Characteristics of Naphthalenediimide-Based Co-Polymers with Different Oligothiophene Donor Units. Adv. Funct. Mater., in press; DOI: 10.1002/adfm.201302297 (2013).
Li, J., Du, J., Xu, J., Chan, H. L. W. & Yan, F. The influence of gate dielectrics on a high-mobility n-type conjugated polymer in organic thin-film transistors. Appl. Phys. Lett. 100, 033301–033304 (2012).
Caironi, M. et al. Very Low Degree of Energetic Disorder as the Origin of High Mobility in an n-channel Polymer Semiconductor. Adv. Funct. Mater. 21, 3371 (2011).
Yan, H., Schuettfort, T., Kronemeijer, A. J., McNeill, C. R. & Ade, H. W. Influence of dielectric-dependent interfacial widths on device performance in top-gate P(NDI2OD-T2) field-effect transistors. Appl. Phys. Lett. 101, 093308–093304 (2012).
Schuettfort, T. et al. Surface and Bulk Structural Characterization of a High-Mobility Electron-Transporting Polymer. Macromolecules 44, 1530–1539 (2011).
Takacs, C. J. et al. Remarkable Order of a High-Performance Polymer. Nano Lett. 13, 2522–2527 (2013).
Giussani, E., Fazzi, D., Brambilla, L., Caironi, M. & Castiglioni, C. Molecular Level Investigation of the Film Structure of a High Electron Mobility Copolymer via Vibrational Spectroscopy. Macromolecules 46, 2658–2670 (2013).
Rivnay, J. et al. Unconventional face-on texture and exceptional in-plane order of a high mobility n-type polymer. Adv. Mater. 22, 4359–4363 (2010).
Rivnay, J. et al. Drastic Control of Texture in a High Performance n-Type Polymeric Semiconductor and Implications for Charge Transport. Macromolecules 44, 5246–5255 (2011).
Schuettfort, T., Thomsen, L. & McNeill, C. R. Observation of a Distinct Surface Molecular Orientation in Films of a High Mobility Conjugated Polymer. J. Am. Chem. Soc. 135, 1092–1101 (2013).
Yang, H. et al. Effect of Mesoscale Crystalline Structure on the Field-Effect Mobility of Regioregular Poly(3-hexyl thiophene) in Thin-Film Transistors. Adv. Funct. Mater. 15, 671–676 (2005).
Crossland, E. J. W., Rahimi, K., Reiter, G., Steiner, U. & Ludwigs, S. Systematic Control of Nucleation Density in Poly(3-Hexylthiophene) Thin Films. Adv. Funct. Mater. 21, 518–524 (2011).
Steyrleuthner, R. et al. Aggregation in a High-Mobility n-Type Low-Bandgap Copolymer with Implications on Semicrystalline Morphology. J. Am. Chem. Soc. 134, 18303–18317 (2012).
Facchetti, A. Organic semiconductors: Made to order. Nat. Mater. 12, 598–600 (2013).
Sciascia, C. et al. Sub-Micrometer Charge Modulation Microscopy of a High Mobility Polymeric n-Channel Field-Effect Transistor. Adv. Mater. 23, 5086 (2011).
Wang, L., Fine, D., Basu, D. & Dodabalapur, A. Electric-field-dependent charge transport in organic thin-film transistors. J. Appl. Phys. 101, 054515 (2007).
Caironi, M. et al. Efficient charge injection from a high work function metal in high mobility n-type polymer field-effect transistors. Appl. Phys. Lett. 96, 183303–183303 (2010).
Brown, P. J., Sirringhaus, H., Harrison, M., Shkunov, M. & Friend, R. H. Optical spectroscopy of field-induced charge in self-organized high mobility poly(3-hexylthiophene). Phys. Rev. B 63, 125204 (2001).
Sze, S. M. & Ng, K. K. Physics of Semiconductor Devices, (3rd Ed., John Wiley & Sons, Inc. 2006).
Acknowledgements
The authors are grateful to Nicola Martino, Sadir Bucella and Dr. Annamaria Petrozza for assisting at different stages the research and to Dario Natali for useful discussions during the revision of the manuscript. Partial financial support is acknowledged from Fondazione Cariplo under project Indixi, Grant No. 2011-0368 and from the European Union through the Marie-Curie Career Integration Grant 2011“IPPIA”, within the EU Seventh Framework Programme (FP7/2007–2013) under grant agreement No. PCIG09-GA-2011-291844.
Author information
Authors and Affiliations
Contributions
A.L. and M.C. conceived the research and devised the experiments. A.L. prepared all the samples for electrical, electro-optical and AFM characterization and performed electrical and AFM measurements. A.L. and L.C. prepared samples for optical characterization and performed polarized microscopy experiments. V.D. performed charge-modulation spectroscopy measurements. A.L., L.C. and M.C. interpreted the data and co-wrote the paper. All the authors assisted in the preparation and in the editing of the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Reprints and permission information is available online at http://npg.nature.com/reprintsandpermissions/
Electronic supplementary material
Supplementary Information
Supplementary Information
Supplementary Information
Optical Microscope View of birefringent films at different polarisation angles
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Luzio, A., Criante, L., D'Innocenzo, V. et al. Control of charge transport in a semiconducting copolymer by solvent-induced long-range order. Sci Rep 3, 3425 (2013). https://doi.org/10.1038/srep03425
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep03425
This article is cited by
-
A critical review on 4D printing and their processing parameters
International Journal on Interactive Design and Manufacturing (IJIDeM) (2023)
-
Chiral emergence in multistep hierarchical assembly of achiral conjugated polymers
Nature Communications (2022)
-
Foundry-compatible high-resolution patterning of vertically phase-separated semiconducting films for ultraflexible organic electronics
Nature Communications (2021)
-
n-Type D-A Conjugated Polymers: Relationship Between Microstructure and Electrical/Mechanical Performance
Chemical Research in Chinese Universities (2021)
-
Microstructural control suppresses thermal activation of electron transport at room temperature in polymer transistors
Nature Communications (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
