
Abstract
The anti-apoptotic protein Bax inhibitor-1 (BI-1) is a regulator of apoptosis linked to endoplasmic reticulum (ER) stress. It has been hypothesized that BI-1 protects against neuron degenerative diseases. In this study, BI-1−/− mice showed increased vulnerability to chronic mild stress accompanied by alterations in the size and morphology of the hippocampi, enhanced ROS accumulation and an ER stress response compared with BI-1+/+ mice. BI-1−/− mice exposed to chronic mild stress showed significant activation of monoamine oxidase A (MAO-A), but not MAO-B, compared with BI-1+/+ mice. To examine the involvement of BI-1 in the Ca2+-sensitive MAO activity, thapsigargin-induced Ca2+ release and MAO activity were analyzed in neuronal cells overexpressing BI-1. The in vitro study showed that BI-1 regulates Ca2+ release and related MAO-A activity. This study indicates an endogenous protective role of BI-1 under conditions of chronic mild stress that is primarily mediated through Ca2+-associated MAO-A regulation.
Similar content being viewed by others


Introduction
Bax inhibitor-1 (BI-1) is an evolutionarily conserved, multi-transmembrane protein that is located in the endoplasmic reticulum (ER) and provides cytoprotective functions in both animals and plants1,2. The cytoprotective function of BI-1 was originally discovered in cDNA library screens for human proteins capable of suppressing the death induced by ectopic expression of mammalian Bax protein in yeast3. When overexpressed, the anti-apoptotic protein BI-1 has been shown to reduce ER Ca2+ concentrations, possibly through a mechanism involving inositol 1,4,5-trisphosphate receptor (IP3R) activation4,5. Knockdown of BI-1 results in increased ER basal Ca2+ concentration and increased Ca2+ release into the cytosol following thapsigargin challenge4. The regulatory role of BI-1 in Ca2+ may contribute to its neuroprotective effects. As such, BI-1 has recently been identified as a critical constituent in both ischemia and traumatic brain injury mouse models, showing regulatory effects against serotonin-depletion stress6,7. Disturbances in intracellular Ca2+ homeostasis have been reported in depression and bipolar diseases8,9. These disruptions hinder cell viability and function. Although BI-1 also confers resilience in animal models7,10, the mechanism by which BI-1 protects against depression is not clearly understood.
Neuronal Ca2+ is linked both directly and indirectly to monoamine oxidases (MAOs), the core enzymes for monoamine metabolism. The MAOs, which are located in the mitochondrial membrane, metabolize monoamines and contribute to the maintenance of monoamine homeostasis11. Biochemical and pharmacological studies have revealed two isoforms of MAO, MAO-A and B12,13. While MAO-A oxidizes serotonin, norepinephrine and dopamine, MAO-B preferentially oxidizes dopamine. Ca2+ accumulation leads to enhanced MAO activity in neuronal cells and neuronal disease models14,15.
In this study, we tested the hypothesis that BI-1 is involved in Ca2+-related psychiatric conditions including depression through endogenous Ca2+ homeostatic regulation. This study linked MAO-A activity with BI-1-associated Ca2+ regulation in a chronic mild stress model.
Results
BI-1 mice are more vulnerable to stress-induced depression-like behavior than BI-1+/+ mice
BI-1+/+ and BI-1−/− mice aged 7 to 8 months were exposed to chronic mild stress for 2 or 6 weeks according to the experimental design (Supplemental Fig. 1). During the stress procedures, body weight was significantly more decreased in BI-1−/− mice than in BI-1+/+ mice (Supplementary Fig. 2). Because corticosteroid is considered to be a stress marker16,17, the level of serum corticosterone was compared between BI-1+/+ and BI-1−/− mice with or without stress. In BI-1−/− mice, the corticosteroid concentration was greatly increased under conditions of stress compared with BI-1+/+ mice, especially in the 6-weeks group, with the BI-1−/− group showing high sensitivity to stress (Fig. 1A). Sucrose intake, another representative marker for the stress response, was significantly decreased in the 6-weeks stress group of BI-1−/− mice compared with BI-1+/+ mice (Fig. 1B). The response to stress was also compared between groups through analysis of locomotor activity. Significant differences were observed in distance traveled and locomotor time between the two groups under the 6-weeks chronic mild stress regime (Fig. 1C, Supplementary Table 1); these two parameters were significantly higher in the BI-1−/− mice relative to the wild-type mice, suggesting that knockout of BI-1 increases vulnerability to chronic mild stress. Forced swimming, which imposes high stress and evokes marked changes in physical and emotional components18, had a similar effect on swimming time in both BI-1+/+ and BI-1−/− stressed mice (Supplementary Fig. 3).

BI-1 inhibits chronic mild stress-induced depression.
(A) Serum corticosteroid levels in BI-1+/+ and BI-1−/− mice were measured after 2 and 6 weeks of chronic mild stress. (B) Sucrose consumption of BI-1+/+ and BI-1−/− mice was measured over 6 weeks. (C) Spontaneous locomotor activities including distance traveled (left) and locomotor time (right) were measured after 2 and 6 weeks of chronic mild stress and were significantly different in BI-1−/− mice compared with the BI-1+/+ control at each time point (*p < 0.05).
BI-1 affects changes in hippocampus volume, ROS accumulation, BDNF expression and ER stress associated with chronic mild stress
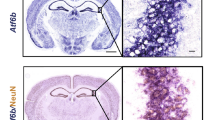
Since the hippocampus is known to be vulnerable to stress that induces depression19, the change in the volume of the hippocampus of BI-1+/+ and BI-1−/− mice was measured by MRI image analysis. Representative MRI images of hippocampi are shown in Figure 2A. Hippocampus volume was quantified and compared between BI-1+/+ and BI-1−/− mice. The volume of the hippocampus in the BI-1 knockout mice was significantly reduced in the 6-weeks stress group (Fig. 2B). In addition, hippocampus structure was analyzed through hematoxylin and eosin (H&E) staining. Although the hippocampal morphology of mice exposed to 2 weeks of stress was similar to that of controls, hippocampus structural alterations were observed after 6 weeks of stress and were more dramatic in the BI-1 knockout mice (Fig. 2C).

BI-1 knock-out enhances hippocampus volume and structure changes and ROS accumulation under chronic mild stress.
(A) Brain images and (B) hippocampus volumes from BI-1+/+ and BI-1−/− mice were analyzed after 6 weeks of chronic mild stress. (C) H&E staining of BI-1+/+ and BI-1−/− mouse brains was performed after 2- and 6-weeks periods of chronic mild stress. (D) Dihydroethidium staining of BI-1+/+ and BI-1−/− mouse brains was performed after 2- and 6-weeks chronic mild stress treatment and the fluorescence intensity was quantified (lower).
Dihydroethidium staining was performed in frozen sections to examine reactive oxygen species (ROS) involvement in the chronic mild stress response. As expected, we observed ROS accumulation over the 2- and 6-weeks stress periods, especially in the BI-1 knockout condition (Fig. 2D, quantified in the lower panel). BI-1+/+ mice showed positive staining for brain-derived neurotrophic factor (BDNF) in the hippocampus, with a decrease in expression during the 6-weeks stress period. Compared with the staining pattern in BI-1+/+ mice, BDNF was not positively identified over the 6-weeks stress condition in BI-1−/− mice, particularly in the dentate gyrus (Fig. 3A). Based on these data, it was necessary to examine the ER stress response, because BI-1 is an established ER stress regulator and ER stress regulation has been suggested as a possible therapeutic mechanism for psychiatric diseases including depression20,21. Indeed, the ER stress response was more clearly observed in the hippocampus following exposure to chronic mild stress in the BI-1−/− mice compared with the BI-1+/+ mice, as demonstrated by immunostaining of representative ER stress response proteins GRP78 and CHOP and cleaved caspase-3 (Fig. 3B). As shown in Figure 3C, GRP78, CHOP, p-IRE-1α and spliced XBP-1 were clearly expressed in the BI-1−/− mice exposed to 6 weeks of chronic mild stress, consistent with vulnerability to stress in the BI-1 knock-out condition. Taken together, these morphological and signaling changes in the hippocampus suggest that stress mainly affects the hippocampus region, where BI-1 regulates ROS accumulation and ER stress leading to alterations in the structure of the hippocampi.

BI-1 knock-out regulates the expression of BDNF, enhancing ER stress response under chronic mild stress.
(A) Immunoblotting and immunostaining with anti-BDNF were performed in BI-1+/+ and BI-1−/− mouse brains after 2- and 6-weeks of chronic mild stress. (B) Immunostaining with anti-GRP78 or anti-CHOP antibody was performed in BI-1+/+ and BI-1−/− mouse brains after 2 and 6 weeks of chronic mild stress. Arrow; positively stained region. (C) Immunoblot analysis of BI-1+/+ and BI-1−/− mouse brain tissues after 2 and 6 weeks of chronic mild stress was performed with anti-GRP78, CHOP, p-eIF-2α, eIF-2α, PERK, IRE-1α, p-IRE-1α, sXBP-1 or β-actin antibodies. All proteins showed a significant difference in expression in BI-1−/− brain compared with the BI-1+/+ control at each time (*p < 0.05). Although cropped blots were used, the gels were run under the same experimental conditions.
BI-1 regulates MAO-A activity, leading to the regulation of serotonin, norepinephrine or dopamine
ER stress is also associated with the disturbance of ER function including the disruption of Ca2+ homeostasis. Since Ca2+ disturbance increases the activity of monoamine oxidase-A (MAO-A), a mitochondria-bound enzyme that generates ROS as a natural by-product of the deamination of neurotransmitters such as serotonin22, MAO-A and MAO-B enzyme activities were analyzed in brains from BI-1+/+ and BI-1−/− mice following exposure to chronic mild stress. In the BI-1−/− mice, the activity of MAO-A, but not MAO-B, was significantly increased, especially in mice exposed to 6 weeks of chronic mild stress (Fig. 4A). MAO-A and MAO-B mRNA and protein expressions were also analyzed in BI-1+/+ and BI-1−/− mice following exposure to chronic mild stress. No differences were found between the two groups of mice (Supplementary Fig. 4A, B). These data suggest that BI-1 affects MAO-A activity post-translationally. The concentrations of representative neurotransmitters were analyzed through LC-MS-MS analysis. Levels of the monoamines serotonin, norepinephrine and dopamine in whole brains from BI-1−/− mice were significantly decreased after 6 weeks of chronic mild stress compared with the BI-1+/+ mice (Fig. 4B, Supplementary Table 2).

BI-1 knock-out enhances MAO-A activity, leading to the regulation of monoamines.
(A) MAO-A and -B activity in BI-1+/+ and BI-1−/− mouse brain tissues was measured after 2 and 6 weeks of chronic mild stress. (B) Serotonin, norepinephrine and dopamine levels in BI-1+/+ and BI-1−/− mouse brain tissues were analyzed after 2 and 6 weeks of chronic mild stress. Levels in BI-1−/− brains were significantly different from those of the BI-1+/+ control at 6 weeks (*p < 0.05).
BI-1 regulates cytosolic Ca2+, leading to the regulation of MAO-A activity
It is known that ER Ca2+ release induced by BI-1 can cause reduced intra-ER Ca2+ concentrations2,5. We generated HT22 hippocampus neuron cells stably expressing BI-1 or a neomycin-resistant vector control (Neo). The stable expression of BI-1 was confirmed by measuring BI-1 production from BI-1 transfected clones #2-, #8- and #12 (Fig. 5A, upper). Neo-resistant (#4) and BI-1-expressing (#2-, #8- and #12) cells were treated with 1 μM thapsigargin to examine the effects of increased Ca2+ availability on MAO-A activity in living cells. Using the cells stably expressing BI-1, we were able to investigate the regulatory effects of BI-1 on cytosolic Ca2+ concentration. Although a low level of expression of BI-1 (clone #2) slightly inhibited the Ca2+ spike from thapsigargin compared with Neomycin-resistant vector-transfected (Neo) cells, higher expression of BI-1 (clone #12) significantly inhibited thapsigargin-induced Ca2+ stimulation compared with Neo cells. In Figure 5B, higher concentrations of thapsigargin (2 and 5 μM) were used to treat the cells. Compared with the Ca2+ increase in Neo cells, the Ca2+ increase was significantly regulated in the BI-1 cells compared with Neo cells and was more significantly regulated in the cells with the highest expressions of BI-1 (#12). The relationship between MAO-A activity and intracellular Ca2+ was then analyzed. The thapsigargin-treated BI-1 cells exhibited low levels of MAO-A activity relative to the levels observed in Neo cells (Fig. 5C). BI-1#12 cells were chosen to explore MAO-A activity because of their high level of BI-1 expression. ROS accumulation was examined through DCF-DA staining analyses. A smaller amount of ROS accumulated in thapsigargin-treated BI-1#12 cells compared with control cells (Fig. 5D). Expression of the MAO-A gene (expressed as a ratio to GAPDH) was not affected by thapsigargin treatment (Supplementary Fig. 4A). MAO-A protein expression was also unchanged compared with control levels (Supplementary Fig. 4B), indicating that Ca2+ affects MAO-A activity, but not its transcription or translation.

BI-1 regulates Ca2+, leading to the regulation of MAO-A activity.
(A) Immunoblotting was performed with anti-HA or anti-β-actin antibody in Neo and BI-1 transfected cells (clones Neo #4, #15 or #34 and BI-1 #2, #8 or #12). Transfected cells were treated with 1 μM thapsigargin and Ca2+ was analyzed as described in the Materials and Methods (lower). (B) Neo and BI-1 expressing clones #2, #8 or #12 cells (expressing difference levels of BI-1) were treated with 2 or 5 μM thapsigargin and Ca2+ was analyzed and quantified. Ca2+ levels in BI-1 cells were significantly different from those in Neo cells for treatment with 2 μM thapsigargin (*p < 0.05) or 5 μM thapsigargin (#p < 0.05). (C) Neo and BI-1 cells were treated with 1, 2 or 5 μM thapsigargin for 30 min and the MAO activity was measured as described in Materials and Methods. (D) Neo and BI-1 cells were treated with 1, 2 or 5 μM thapsigargin for 12 hours and ROS was measured as described in Materials and Methods. ROS levels in BI-1 cells were significantly different from those in Neo cells for each condition (*p < 0.05).
Discussion
The findings of this study demonstrated the endogenous role of BI-1 in resilience against stress and suggested that BI-1 exhibits Ca2+-associated MAO-A regulatory activity. BI-1−/− mice showed vulnerability to chronic mild stress with alterations in hippocampus size and morphology and enhanced ROS accumulation and ER stress response. MAO-A was highly activated and the concentrations of monoamines including serotonin, norepinephrine and dopamine were reduced, especially following chronic exposure to mild stress. Through an in vitro study, we showed that BI-1 regulates Ca2+ release and MAO-A activity, consistent with the in vivo results.
Based on our study, BI-1 gene deficiency is implicated in depression-like behaviors during chronic mild stress. The BI-1−/− stress animals consumed smaller amounts of sucrose than the BI-1+/+ stress group (Fig. 1B), suggesting that BI-1 gene deficiency is involved in the induction of anhedonic-like behavior by mild stress. Compared with the BI-1+/+ stress group, the BI-1−/− stress group showed a significant increase in spontaneous locomotor activity and immobility time (Fig. 1C), suggesting that BI-1 deficiency causes enhanced vulnerability to chronic mild stress. However, it should be noted that younger mice (13 weeks) showed a decrease in only sugar consumption, but not locomotor activities, in BI-1 knockout mice compared with wild-type mice during 1 or 2 weeks of treatment10. There may be an age-dependent vulnerability to chronic stress because the animals used in our model were 7–8 months old, relatively older than those used in the previous study. Consistent with our results, the elderly are more susceptible to stress conditions including psychological stress23. Oxidative stress has been implicated in a number of age-related degenerative processes in the central nervous system24,25. Why the incidence of chronic disease, including psychiatric diseases, increases with age is still an unanswered question; however, the role of ER stress has recently been a focus in studies of aging processes26,27,28. The present study suggests that older age may be related to high susceptibility to chronic stress, especially in the BI-1 knockout condition, most likely due to alterations in ER function and ER stress responses.
The BI-1 protein has recently been established as a regulator of ER stress29,30. The ER is a critical site for intracellular Ca2+ storage as well as protein synthesis, folding and trafficking. ER stress results in cell death by causing a disturbance of Ca2+ homeostasis and leading to activation of caspase-331. ER stress is involved in neurodegenerative diseases such as Parkinson's disease, Alzheimer's disease, cerebral ischemia32,33,34,35,36 and depression37. Notably, certain antidepressants modulate the ER stress response and Ca2+ homeostasis. Many antidepressants such as fluvoxamine, a selective serotonin-reuptake inhibitor and imipramine, a tricyclic antidepressant, regulate ER stress37. Without the endogenous ER stress regulator gene BI-1, an increase in active caspase-3 was detected under chronic mild stress, indicating induction of the ER stress response (Fig. 3B, C). Similarly, overexpressing BI-1 regulates ER stress through the Ca2+ buffering capacity of the ER and has been shown to be neuroprotective in several studies2,6,38. More recently, a transgenic mouse model with BI-1 expression under the control of the neuron-specific enolase promoter showed that BI-1 plays a preventive role against chemical serotonin deprivation stress7, although this model did not show any positive ER stress phenomena. However, the results of the study suggested the ability of BI-1 to maintain Ca2+ homeostasis and contribute to the preventive/regulatory effect against serotonin deprivation. Homeostatic control of ER Ca2+ is essential for appropriate ER function, including protein folding. Elevated cytosolic Ca2+ levels have been associated with apoptosis and necrosis in neuronal cultures39,40 and this Ca2+ disruption may underlie pathologic mechanisms of psychiatric disorders41. Considering the pathologic roles of Ca2+ disturbance, BI-1-associated Ca2+ maintenance may contribute to controlling ER stress-associated progression of depression.
In the BI-1 knockout condition, MAO-A activity induced by chronic stress was significantly higher than that of BI-1 wild-type groups (Fig. 4A), which was correlated with monoamine concentrations (Fig. 4B). In Neo cells, Ca2+ levels increased after treatment with thapsigargin, a Ca2+-ATPase inhibitor. The Ca2+ increase was smaller in the thapsigargin-treated BI-1 cells compared with the Neo cells (Fig. 5A). This finding also correlated with MAO-A activity in the thapsigargin-treated condition (Fig. 5C). The requirement of Ca2+ for MAO-A activity has recently been demonstrated ex vivo in the brain42,43,44 and is supported by the ability of the Ca2+-channel antagonist nimodipine to block the selective increase in MAO-A activity observed in senescence-accelerated mouse brains45. Consistent with this, the Ca2+-dependent processes and pathology of encephalopathic brains are linked to a selective increase in MAO-A activity46. It has also been suggested that increased intracellular free Ca2+ levels and associated MAO-A activity contribute to the oxidative stress associated with neuron degenerative diseases47,48. MAO-A is a risk factor in neuron degenerative diseases such as Alzheimer's disease. In BI-1 knockout mice, ROS accumulated during catecholamine metabolism due to chronic mild stress (Fig. 2D). Consistent with this, ROS accumulation was much lower in the thapsigargin-treated BI-1-overexpressing neuronal cells compared with control cells (Fig. 5D). Given that 5-HT is metabolized by MAOs and H2O2 is generated in the process49, the lack of BI-1-associated enhancement of MAO-A activity may increase 5-HT metabolism, resulting in the accumulation of H2O2. The mitochondria-bound enzyme MAO generates peroxyradicals as a natural by-product of the deamination of neurotransmitters such as serotonin16. Changes in MAO-A activity parallel changes in the production of ROS such as H2O242. Our results also suggest that the oxidative stress associated with chronic mild stress could be due to Ca2+-sensitive MAO-A-mediated phenomena, which are regulated by the presence of BI-1.
In conclusion, our findings provide mechanistic support for the hypothesis that BI-1 targets ER Ca2+ homeostasis to regulate monoamines including serotonin, norepinephrine and dopamine. These findings hold exciting promise for the development of new treatments for mood disorders. New treatments that specifically target neuronal BI-1 and its regulated pathways may provide innovative mechanisms to enhance brain plasticity in patients with psychiatric disease.
Methods
Reagents and antibodies
5-Hydroxytryptamine (5-HT), β-phenylethylamine (PEA), the β-actin antibody and protease inhibitor cocktail were bought from Sigma-Aldrich Co. [14C]-5-HT (NEC-225) and [14C]-PEA (NEC-502) were purchased from PerkinElmer Life Sciences. Phosphate-buffered saline (PBS) was purchased from Invitrogen (Carlsbad, CA). Serotonin, dopamine and norepinephrine were obtained from Sigma (St Louis, MO). Water was purified by a Milli-Q system from Millipore (Bedford, MA). LC-MS grade acetonitrile and formic acid were from Fischer Scientific (Fair Lawn, NJ). All other chemicals were of analytical grade and were purchased from Sigma.
Animals
BI-1−/− and BI-1+/+ C57BL female mice (n = 33 each, 7–8 months old) were kindly provided by Dr. John C. Reed of the Stanford-Burnham Institute for Medical Research (La Jolla, CA, USA) and housed in groups of 3 to 5 animals at 21 ± 1°C and 55 ± 5% humidity under a 12-hours/12-hours light/dark cycle. Mice were maintained in specific pathogen-free housing and were cared for in accordance with the National Institutes of Health Guidelines for Animal Care. The Animal Care and Use Committee of Chonbuk National University Graduate School of Medicine approved this study (Approval number: CBU 2013-0013).
Study design for the chronic mild stress procedure
The procedure for inducing chronic mild stress (CMS) was based on a protocol described previously10,50,51, but with a difference in mouse age. Briefly, the CMS procedure was applied to each mouse and consisted of a variety of unpredictable mild environmental, social and physical stressors, including confinement in a small tube (1 hours), an empty cage without sawdust (15 hours), water and food deprivation (15 hours), food restriction (approximately 50 mg of food pellets, 3 hours), cage tilted at 45° (1–3 hours), damp sawdust (approximately 200 ml of water per 100 g of sawdust, 3 hours), paired housing in damp sawdust (18 hours), reversal of the light/dark cycle, 4-hour light/dark succession every 30 minutes (09:30–11:30) and light (15–17 hours) dark (4–6 hours). Control animals were single-housed during the time of CMS to match the condition, but otherwise were group-housed in a different room and left undisturbed with the exception of general handling (e.g., regular cage cleaning and measuring body weight). Female mice aged 7 to 8 months were assigned to eight different groups: BI-1+/+ control and stress groups (n = 8–9) and BI-1−/− control and stress groups (n = 8–9) for either a 2- or 6-weeks period.
Sucrose consumption test
Sucrose intake was measured in the control and stress groups once per weeks on separate days during a 15-hours window (18:00–09:00) following 8 hours of food and water deprivation as previously described10. Consumption was measured by weighing the bottle at the beginning and end of the test. Intake was expressed in relation to animal body weight (g/g).
Spontaneous locomotor activity
Locomotor activity was measured in an open field box (40 × 50 × 30 cm) consisting of a white base and black walls using a Smart video tracking system (Panlab S.L., Barcelona, Spain) in a softly illuminated (40 W) soundproof room. Each mouse was placed in the center of the open field apparatus and the distance traveled (cm), locomotor time (seconds), number of times crossing the center (50% of all area) and time spent in the center were recorded at 10-min intervals during a 30-min period. The open field arena was thoroughly cleaned between tests.
Magnetic resonance imaging measurements
A total of 124 sagittal T1-weighted (TR = 24, TE = 5, FA = 45) gradient echo images were obtained using a 1.5 GE Sigma imager. The MRI volumetric method used was similar to that previously reported. Sagittal images were acquired with a 24-cm field of view using a slice thickness of 1.2 mm without gaps and a 256 × 192 matrix. For anatomical work, scans were reformatted at a 2-mm slice thickness in the coronal plane. Using the Multimodal Image Data Analysis System (MIDAS) image analysis software (Tsui, M.H. MIDAS Version 1.0, unpublished manual, 1995) developed in-house at NYU with a Sun Sparc workstation (Sun Microsystems, Mountain View, California), hippocampus regions were drawn in three-fold enlarged images. All of the image analyses were performed blind to group membership. Tissue volumes were estimated by counting the number of remaining parenchymal pixels (of known size) over the slices measured. The hippocampus and head size were sampled every 2 mm; in most cases this included 12 to 15 coronal sections.
Dihydroethidium staining
Frozen and enzymatically intact sections (30-mm thick) were prepared from mouse brains and immediately incubated with dihydroethidium (10 mol/L; Probes Inc., Eugene, OR, USA) in phosphate-buffered saline for 30 min at 37°C in a humidified chamber. Dihydroethidium is oxidized on reaction with superoxide to ethidium, which binds to DNA in the nucleus and fluoresces red. Images were observed using a fluorescence microscope (PROVISAX80; Olympus). The intensity of the fluorescence was analyzed and quantified using Image J.
Monoamine oxidase activity
MAO-A or B activity (nmol/hours/mg protein) in BI-1+/+ or BI-1−/− mice and Neomycin resistant vector or BI-1-stably transfected HT-22 cells were measured radiochemically, as previously described42. Briefly, for the MAO-A and MAO-B assay, the frozen control or stressed BI-1+/+ and BI-1−/− mouse brain samples were diluted 1:3 (vol/vol) with 50 mM potassium phosphate buffer pH 7.5. Aliquots of 100 μL mouse brain homogenate and 250 μL 50 mM phosphate buffer pH 7.5 were preincubated in a 37°C water bath for 1 min. The enzyme reaction was started by the addition of 150 μL of [14C]5-hydroxytryptamine solution (0.03 μCi) for MAO-A and 50 μL [14C]phenylethylamine solution (0.01 μCi) for MAO-B. After incubating the samples for exactly 15 min at 37°C, the reaction was terminated by addition of 100 μL 4 M HCl. The [14C]-labeled deaminated reaction products 5-hydroxyindolacetic acid and phenylacetic acid were extracted with 5 mL toluene/ethylacetate (1:1) by shaking for 10 min on a horizontal shaker (Edmund Bühler SM25, Tübingen, Germany). After centrifugation at 100 × g for 5 min, the aqueous bottom phase was frozen at −70°C for 15 min and the organic phase was decanted into counting vials. After the addition of 5 mL scintillation liquid, the radioactivity was measured by a liquid scintillation counter. The results were calculated as pmol deaminated metabolite, 5-hydroxyindolacetic acid and phenylacetic acid formed per min. In both assays, protein content was measured using bovine serum albumin as a standard. For transfected HT-22 cells, the cells were incubated with 1, 2 or 5 μM thapsigargin for 30 min and then washed with cold PBS and homogenized with 50 mM potassium phosphate buffer pH 7.5. An aliquot (100 μg/100 μL) was incubated with 150 μL of [14C]5-hydroxytryptamine solution (0.03 μCi) for MAO-A and 50 μL [14C]phenylethylamine solution (0.01 μCi) for MAO-B for 10 min at 37°C. All the other processes were same as for the MAO activity analysis for the brain tissues described above.
Monoamine analysis
Quantification of dopamine, norepinephrine and serotonin in brain samples was conducted by HPLC coupled with mass spectrometry. HPLC separation was performed using an Agilent 1100 system (Agilent, Palo Alto, CA, USA). Chromatography separation was performed using a ZORBAX SB-Phenyl Narrow Bore column (150 × 2.1-μm internal diameter and 5-μm particle size). Dopamine, norepinephrine and serotonin were separated by gradient elution. The mobile phase was composed of solvent A (water) and solvent B (acetonitrile) and both solvents contained 0.1% formic acid. The gradient run started with 5% solvent B for 1 min and increased to 100% over 20 min, where it remained for another minute before returning to 5% over 25 min, which was maintained for 7 min. The total run time was 32 min at a flow rate of 0.2 mL/min. An Agilent Technologies 6410 triple quadrupole mass spectrometer equipped with electrospray ionization (ESI) in the positive ionization mode was used. The ESI-MS/MS parameters were set as follows: gas temperature 300°C, gas flow 10 L/min and capillary voltage 4 kV. Samples were analyzed by multiple reactions monitoring (MRM) and the MRM transitions used were m/z 177–160 for serotonin, m/z 170–152.1 for norepinephrine and m/z 154.2–137.1 for dopamine. The fragmentation voltages used were 30 V for dopamine and 50 V for serotonin and norepinephrine. The collision energy was set at 3 eV for norepinephrine and 5 eV for serotonin and dopamine. Data acquisition was performed with MassHunter software.
Cell culture
The immortalized mouse hippocampal HT-22 cell line was obtained from the American Type Cell Collection (ATCC). The cells were stably transfected with the Neomycin-resistant pcDNA3 (Neo) or pcDNA3-BI-1-HA vector (BI-1) using the SuperFect transfection reagent (Qiagen, Crawley, UK). The cells were cultured for 5 weeks in 1 mg/mL G418 (Invitrogen, Carlsbad, CA, USA). Cells were maintained in DMEM/low glucose medium containing 10% fetal bovine serum, 100 IU/mL penicillin G sodium salt and 0.03% glutamine at 37°C and 5% CO2.
Calcium analysis
The procedures for Ca2+ measurement were modified from Kim et al.5. Briefly, the low affinity fluorescent Ca2+ dye Fura-2/AM (1-[2-(5-carboxyoxazol-2-yl)-6-aminobenzoFURAn-5-oxy]-2-(2-amino-5-methylphenoxy)-ethane-N, N, N′, N′-tetraacetic acid pentaacetoxymethyl ester; Molecular Probes, Eugene, OR, USA) was used to measure changes in intracellular (cytosolic) free Ca2+ ([Ca2+]i). Cells were incubated with Fura-2/AM (2 μM) for 30 min at room temperature in Hanks' balanced salt solution. After loading, cells were washed three times in isotonic buffer without Ca2+ (KH buffer: 132 mM NaCl, 5 mM KCl, 10 mM glucose, 10 mM HEPES and 1.05 mM MgCl2). Cells were then promptly treated with thapsigargin. Changes in [Ca2+]i were determined as a ratio of 340/380-nm excitation (512-nm emission) using an integrated spectrofluorometer (Photon Technology International, Birmingham, NJ, USA). Ca2+ concentrations were calculated using the equation [Ca2+]i = Kd(F380 max/F380 min)(R − Rmin)/(Rmax − R); a Kd value of 229 nM was assumed for the binding of calcium to Fura-2/AM. Rmax and Rmin were determined in each experimental group by the consecutive addition of 30 μM Triton X-100 (Rmax) and 50 mM EGTA (Rmin). Separately, to determine the relative Ca2+ increase, cytosolic Ca2+ was compared between Neo cells and BI-1 cells as a percentage based upon the area of the cytosolic Ca2+ increase curve for Neo cells treated with 2 μM thapsigargin (considered as 100%).
Dichlorofluorescein diacetate (DCF-DA) analysis
The intracellular ROS levels were measured as described previously30. Briefly, 1.5 × 106 cells were incubated with 1, 2 or 5 μM thapsigargin for 12 hours and then treated with 1 μM DCF-DA at 37°C for 30 min. After cooling on ice, the cells were washed with cold phosphate-buffered saline (PBS), scraped from the plate and resuspended in PBS containing 10 mM EDTA. The fluorescence intensity of the 2,7′-dichlorofluorescein formed by a reaction between DCF-DA and the intracellular ROS was analyzed for more than 10,000 viable cells from each sample by PAS flow cytometry (Partec, Münster, Germany) at excitation and emission wavelengths of 488 and 525 nm, respectively. The data were collected and analyzed using Partec software. All experiments were repeated at least eight times, with similar results. The data were expressed as representative histograms from eight independent experiments.
Statistical analysis
All data are expressed as the mean ± standard error of the mean. Body weight and sucrose consumption were analyzed with a repeated-measures two-way analysis of variance (ANOVA). Individual measures of body weight, sucrose consumption and locomotor activity were compared among the groups using a one-way ANOVA followed by Tukey's post hoc test. Differences of p < 0.05 were considered significant.
References
Chae, H. J. et al. Evolutionarily conserved cytoprotection provided by Bax Inhibitor-1 homologs from animals, plants and yeast. Gene 323, 101–13 (2003).
Chae, H. J. et al. BI-1 regulates an apoptosis pathway linked to endoplasmic reticulum stress. Mol Cell 15, 355–66 (2004).
Xu, Q. & Reed, J. C. Bax inhibitor-1, a mammalian apoptosis suppressor identified by functional screening in yeast. Mol Cell 1, 337–46 (1998).
Xu, C., Xu, W., Palmer, A. E. & Reed, J. C. BI-1 regulates endoplasmic reticulum Ca2+ homeostasis downstream of Bcl-2 family proteins. J Biol Chem 283, 11477–84 (2008).
Kim, H. R. et al. Bax Inhibitor-1 Is a pH-dependent regulator of Ca2+ channel activity in the endoplasmic reticulum. J Biol Chem 283, 15946–55 (2008).
Krajewska, M. et al. Endoplasmic reticulum protein BI-1 modulates unfolded protein response signaling and protects against stroke and traumatic brain injury. Brain Res 1370, 227–37 (2011).
Hunsberger, J. G. et al. Bax inhibitor 1, a modulator of calcium homeostasis, confers affective resilience. Brain Res 1403, 19–27 (2011).
Aldenhoff, J. B., Dumais-Huber, C., Fritzsche, M., Sulger, J. & Vollmayr, B. Altered Ca(2+)-homeostasis in single T-lymphocytes of depressed patients. J Psychiatr Res 31, 315–22 (1997).
Kato, T. Molecular neurobiology of bipolar disorder: a disease of ‘mood-stabilizing neurons’? Trends Neurosci 31, 495–503 (2008).
Sui, Z. Y. et al. Effects of chronic mild stress in female bax inhibitor-1-gene knockout mice. Clin Psychopharmacol Neurosci 10, 155–62 (2012).
Youdim, M. B. & Finberg, J. P. New directions in monoamine oxidase A and B selective inhibitors and substrates. Biochem Pharmacol 41, 155–62 (1991).
Squires, R. F. Multiple forms of monoamine oxidase in intact mitochondria as characterized by selective inhibitors and thermal stability: a comparison of eight mammalian species. Adv Biochem Psychopharmacol 5, 355–70 (1972).
Youdim, M. B. & Riederer, P. Monoamine oxidase B; a misunderstood and misjudged enzyme. Pharmacol Res Commun 20 Suppl 4, 9–14 (1988).
Sandri, G., Panfili, E. & Ernster, L. Hydrogen peroxide production by monoamine oxidase in isolated rat-brain mitochondria: its effect on glutathione levels and Ca2+ efflux. Biochim Biophys Acta 1035, 300–5 (1990).
Alzghoul, L. et al. Altered cerebellar organization and function in monoamine oxidase A hypomorphic mice. Neuropharmacology 63, 1208–17 (2012).
Poon, C. C. et al. Mitochondrial monoamine oxidase-A-mediated hydrogen peroxide generation enhances 5-hydroxytryptamine-induced contraction of rat basilar artery. Br J Pharmacol 161, 1086–98 (2010).
Abazyan, B. et al. Prenatal interaction of mutant DISC1 and immune activation produces adult psychopathology. Biol Psychiatry 68, 1172–81 (2010).
Avgustinovich, D. F., Kovalenko, I. L. & Koryakina, L. A. Effects of single episodes of severe stress on the behavior of male and female CBA/Lac and C57BL/6J mice. Neurosci Behav Physiol 37, 731–7 (2007).
Lupien, S. J. et al. Cortisol levels during human aging predict hippocampal atrophy and memory deficits. Nat Neurosci 1, 69–73 (1998).
Gold, P. W., Licinio, J. & Pavlatou, M. G. Pathological parainflammation and endoplasmic reticulum stress in depression: potential translational targets through the CNS insulin, klotho and PPAR-gamma systems. Mol Psychiatry 18, 154–65 (2013).
Pavlovsky, A. A. et al. Psychological stress, cocaine and natural reward each induce endoplasmic reticulum stress genes in rat brain. Neuroscience 246, 160–9 (2013).
Mickey, B. J. et al. Monoamine oxidase A genotype predicts human serotonin 1A receptor availability in vivo. J Neurosci 28, 11354–9 (2008).
Toth, E. et al. Age-dependent effects of chronic stress on brain plasticity and depressive behavior. J Neurochem 107, 522–32 (2008).
Staecker, H., Zheng, Q. Y. & Van De Water, T. R. Oxidative stress in aging in the C57B16/J mouse cochlea. Acta Otolaryngol 121, 666–72 (2001).
Sohal, R. S. & Weindruch, R. Oxidative stress, caloric restriction and aging. Science 273, 59–63 (1996).
Naidoo, N. ER and aging-Protein folding and the ER stress response. Ageing Res Rev 8, 150–9 (2009).
Chen, G., Gong, M., Yan, M. & Zhang, X. Sevoflurane induces endoplasmic reticulum stress mediated apoptosis in hippocampal neurons of aging rats. PLoS One 8, e57870 (2013).
Sterlemann, V. et al. Chronic social stress during adolescence induces cognitive impairment in aged mice. Hippocampus 20, 540–9 (2010).
Sano, R. et al. Endoplasmic reticulum protein BI-1 regulates Ca(2)(+)-mediated bioenergetics to promote autophagy. Genes Dev 26, 1041–54 (2012).
Lee, G. H. et al. Bax inhibitor-1 regulates endoplasmic reticulum stress-associated reactive oxygen species and heme oxygenase-1 expression. J Biol Chem 282, 21618–28 (2007).
Leem, J. & Koh, E. H. Interaction between mitochondria and the endoplasmic reticulum: implications for the pathogenesis of type 2 diabetes mellitus. Exp Diabetes Res 2012, 242984 (2012).
Arduino, D. M., Esteves, A. R., Cardoso, S. M. & Oliveira, C. R. Endoplasmic reticulum and mitochondria interplay mediates apoptotic cell death: relevance to Parkinson's disease. Neurochem Int 55, 341–8 (2009).
Lindholm, D., Wootz, H. & Korhonen, L. ER stress and neurodegenerative diseases. Cell Death Differ 13, 385–92 (2006).
Hara, H. Involvement of endoplasmic reticulum stress in pathogenesis after cerebral ischemia. Rinsho Shinkeigaku 50, 881 (2010).
Oida, Y., Shimazawa, M., Imaizumi, K. & Hara, H. Involvement of endoplasmic reticulum stress in the neuronal death induced by transient forebrain ischemia in gerbil. Neuroscience 151, 111–9 (2008).
Oono, K. et al. JAB1 participates in unfolded protein responses by association and dissociation with IRE1. Neurochem Int 45, 765–72 (2004).
Hosoi, T., Miyahara, T., Kayano, T., Yokoyama, S. & Ozawa, K. Fluvoxamine attenuated endoplasmic reticulum stress-induced leptin resistance. Front Endocrinol (Lausanne) 3, 12 (2012).
Dohm, C. P. et al. Bax inhibitor-1 protects neurons from oxygen-glucose deprivation. J Mol Neurosci 29, 1–8 (2006).
Liu, D. H. et al. Endogenous nitric oxide induces activation of apoptosis signal-regulating kinase 1 via S-nitrosylation in rat hippocampus during cerebral ischemia-reperfusion. Neuroscience 229, 36–48 (2013).
D'Orsi, B. et al. Calpains are downstream effectors of bax-dependent excitotoxic apoptosis. J Neurosci 32, 1847–58 (2012).
Hough, C., Lu, S. J., Davis, C. L., Chuang, D. M. & Post, R. M. Elevated basal and thapsigargin-stimulated intracellular calcium of platelets and lymphocytes from bipolar affective disorder patients measured by a fluorometric microassay. Biol Psychiatry 46, 247–55 (1999).
Cao, X., Wei, Z., Gabriel, G. G., Li, X. & Mousseau, D. D. Calcium-sensitive regulation of monoamine oxidase-A contributes to the production of peroxyradicals in hippocampal cultures: implications for Alzheimer disease-related pathology. BMC Neurosci 8, 73 (2007).
Samantaray, S., Chandra, G. & Mohanakumar, K. P. Calcium channel agonist, (+/−)-Bay K8644, causes a transient increase in striatal monoamine oxidase activity in Balb/c mice. Neurosci Lett 342, 73–6 (2003).
Kosenko, E. A., Venediktova, N. I. & Kaminskii Iu, G. [Calcium and ammonia stimulate monoamine oxidase A activity in brain mitochondria]. Izv Akad Nauk Ser Biol, 542–6 (2003).
Kabuto, H., Yokoi, I., Mori, A., Murakami, M. & Sawada, S. Neurochemical changes related to ageing in the senescence-accelerated mouse brain and the effect of chronic administration of nimodipine. Mech Ageing Dev 80, 1–9 (1995).
Rao, V. L. Nitric oxide in hepatic encephalopathy and hyperammonemia. Neurochem Int 41, 161–70 (2002).
Kitazawa, M., Anantharam, V. & Kanthasamy, A. G. Dieldrin-induced oxidative stress and neurochemical changes contribute to apoptopic cell death in dopaminergic cells. Free Radic Biol Med 31, 1473–85 (2001).
Burke, W. J., Kristal, B. S., Yu, B. P., Li, S. W. & Lin, T. S. Norepinephrine transmitter metabolite generates free radicals and activates mitochondrial permeability transition: a mechanism for DOPEGAL-induced apoptosis. Brain Res 787, 328–32 (1998).
Girgin Sagin, F., Sozmen, E. Y., Ersoz, B. & Mentes, G. Link between monoamine oxidase and nitric oxide. Neurotoxicology 25, 91–9 (2004).
Ducottet, C., Griebel, G. & Belzung, C. Effects of the selective nonpeptide corticotropin-releasing factor receptor 1 antagonist antalarmin in the chronic mild stress model of depression in mice. Prog Neuropsychopharmacol Biol Psychiatry 27, 625–31 (2003).
Yalcin, I., Aksu, F. & Belzung, C. Effects of desipramine and tramadol in a chronic mild stress model in mice are altered by yohimbine but not by pindolol. Eur J Pharmacol 514, 165–74 (2005).
Acknowledgements
This study was supported by National Research Foundation (2012R1A2A1A3001907 and 2008-0062279) and by Korea Healthcare Technology R&D Project (A121931), Ministry for Health and Welfare, Republic of Korea. We thank Professor Myoung-Goo Kang, at Korea Advanced Institute of Science and Technology, for critical review of this manuscript.
Author information
Authors and Affiliations
Contributions
H.Y.L., G.H.L. and T.R.B. performed experiments and analyzed data; M.A. analyzed monoamines, S.M.L. measured brain micro-CT image, M.R.L. analyzed data, K.Y.J. and K.M.K. analyzed immunostaining, H.J.L. and J.W.L. performed GRP78 and CHOP brain immunostaining, Y.C.C. and Y.C.L. reviewed the manuscript and H.R.K. and H.J.C. designed the research, interpreted data and edited the manuscript. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Lee, HY., Lee, GH., Marahatta, A. et al. The protective role of Bax Inhibitor-1 against chronic mild stress through the inhibition of monoamine oxidase A. Sci Rep 3, 3398 (2013). https://doi.org/10.1038/srep03398
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep03398
This article is cited by
-
Iron Oxide Nanoparticles Affects Behaviour and Monoamine Levels in Mice
Neurochemical Research (2019)
-
Crosstalk Between Endoplasmic Reticulum Stress, Oxidative Stress, and Autophagy: Potential Therapeutic Targets for Acute CNS Injuries
Molecular Neurobiology (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
