
Abstract
The disruption of endoplasmic reticulum (ER) function can lead to neurodegenerative disorders, in which inflammation has also been implicated. We investigated the possible correlation between ER stress and immune function using glial cells. We demonstrated that ER stress synergistically enhanced prostaglandin (PG) E2 + interferon (IFN) γ-induced interleukin (IL)-6 production. This effect was mediated through cAMP. Immune-activated glial cells produced inducible nitric oxide synthase (iNOS). Interestingly, ER stress inhibited PGE2 + IFNγ-induced iNOS expression. Similar results were obtained when cells were treated with dbcAMP + IFNγ. Thus, cAMP has a dual effect on immune reactions; cAMP up-regulated IL-6 expression, but down-regulated iNOS expression under ER stress. Therefore, our results suggest a link between ER stress and immune reactions in neurodegenerative diseases.
Similar content being viewed by others


Introduction
Neurodegenerative diseases have been associated with the accumulation of abnormal proteins, which can trigger cellular stress and inflammation1. Increasing evidence suggests that endoplasmic reticulum (ER) stress is involved in neurodegenerative disorders such as Alzheimer's disease (AD), Parkinson's disease (PD) and cerebral ischemic insults2,3,4,5. However, the link between ER stress and inflammation in neurodegenerative diseases has not yet been clarified in detail. Glial cells, such as astrocytes, microglia and oligodendrocytes, are important cells in the brain and are involved in the immune system. In the present study, we used glial cells as a model to analyze the possible association of ER stress with immune functions.
The ER is an organelle that is responsible for the folding of proteins. When exposed to stress, which perturbs ER function (ER stress), cells activate the unfolded protein response (UPR) to cope. However, when the intensity of the stress is increased to sub-threshold, cells activate an apoptotic pathway. ER stress may be linked with neurodegenerative diseases. Both environmental and genetic factors have been implicated in the pathogenesis of neurodegenerative diseases. Several types of genetic mutations have been linked with these disorders6 and have also been suggested to cause ER stress. Familial as well as cellular models of PD7,8, as well as a familial AD-linked presenilin-1 mutation were shown to be associated with ER stress9. Activation of the UPR has been reported in AD brains10,11 and amyloid β-peptide-induced neuronal cell death was shown to be mediated through ER stress-specific caspases12,13. Interestingly, a recent study suggested that the UPR pathway may be activated downstream of the innate immune system through toll-like receptors (TLRs) in macrophages14. Meanwhile, the prior activation of TLRs was shown to attenuate the activating transcription factor 4 (ATF4)-CCAAT/enhancer-binding protein homologous protein (CHOP) branch of the UPR in macrophages15. These findings indicate a link between ER stress and the immune system in peripheral organs. As well as the peripheral immune system, pro-inflammatory cytokines such as IL-1β, IL-6 and TNF-α are produced and their receptors are expressed in various cell types in the central nervous system (CNS)16,17,18. The increased production of pro-inflammatory cytokines such as IL-1β was shown to induce cyclooxygenase (COX) expression, which activated the synthesis of prostaglandins (PGs) in brain asotrocytes19. In addition, nitric oxide (NO), a short-lived gaseous molecule, was also suggested to be involved in the immune system. NO production is regulated through several types of NO synthase (NOS). Inducible NO synthases (iNOSs) are a family of NOSs, the production of which increases under inflammatory conditions. Cytokines, PGs and NO are mainly produced by glial cells such as astrocytes and microglia under inflammatory conditions and chronic inflammation is a hallmark of neurodegenerative diseases20,21. However, the mechanisms responsible for the regulation of inflammation in neurodegenerative disorders remain unclear. Therefore, we hypothesized that ER stress may affect immune reactions in brain glial cells. We have identified a previously unknown link between ER stress and the immune system. We demonstrated that ER stress enhanced PGE2 + IFNγ-induced IL-6 production, but inhibited iNOS production. These unique dual effects were mediated through cAMP. Therefore, our results suggest a functional link between ER stress and immune reactions in glial cells.
Results
ER stress synergistically enhanced PGE2 + IFNγ-induced IL-6 production
We examined the possible link between ER stress and inflammation by measuring IL-6 levels in primary cultured glial cells. Cells were exposed to tunicamycin (Tm), a potent pharmacologic inducer of ER stress. To determine whether tunicamycin could cause ER stress under the present conditions, glial cells were treated with tunicamycin and analyzed for ER stress-specific UPR-regulated genes. We observed the activation of IRE1α and PERK and the induction of a spliced form of XBP1, CHOP and GRP78 in tunicamycin-treated cells (Fig. 1), which indicated ER stress. We also observed the enhanced expression of IL-6 in tunicamycin-treated cells, which suggested that ER stress can also induce production of IL-6 in glial cells (Fig. 2A). Stimulating glial cells with PGE2 + IFNγ caused a marked increase in IL-6 production (Fig. 2A). Thus, we investigated the effect of ER stress on PGE2 + IFNγ-induced IL-6 production. We observed a synergistic effect on PGE2 + IFNγ-induced IL-6 production following pre-treatment with tunicamycin (Fig. 2A). A similar synergistic effect on PGE2 + IFNγ-induced IL-6 expression was observed at the mRNA level with ER stress (Fig. 2B). Therefore, these effects were regulated at the transcriptional level.

Induction of ER stress in glial cells.
Primary cultured mouse glial cells were treated with a specific ER stress-inducing reagent, tunicamycin (Tm: 1 μg/mL) and analyzed for the activation of UPR-related proteins. The treatment of glial cells with tunicamycin for 5 h caused the activation of IRE1 and PERK. A spliced form of XBP1 was increased 18 h after the tunicamycin treatment. CHOP and GRP78 were increased 24 h after the tunicamycin treatment. Cropped blots were run under the same experimental conditions.

PGE2 + IFNγ- or PGE2-induced IL-6 release was enhanced under ER stress.
(A) Glial cells were treated with tunicamycin (Tm: 1 μg/mL) for 1 h and then stimulated with prostaglandin E2 (PGE2: 15 μM) + interferon γ (IFNγ: 20 ng/mL) for 24 h. IL-6 protein levels were detected by ELISA. ER stress increased PGE2 + IFNγ-induced IL-6 release. ***P < 0.001 PGE2 + IFNγv.s. PGE2 + IFNγ + Tm. n = 5. (B) Glial cells were treated with tunicamycin (Tm: 1 μg/mL) for 1 h and then stimulated with PGE2 (15 μM) + IFNγ (20 ng/mL) for 4 h. IL-6 mRNA levels were detected by RT-PCR. Tm increased PGE2 + IFNγ-induced IL-6 expression. ***P < 0.001 PGE2 + IFNγ v.s. PGE2 + IFNγ + Tm. n = 7. The GAPDH was analyzed as loading control. Full-length gels are presented in Supplementary Fig S2. Full-length gels for GAPDH are presented in Supplementary Fig S3. (C) Glial cells were treated with tunicamycin (Tm: 1 μg/mL) for 1 h and then stimulated with PGE2 (15 μM) for 24 h. IL-6 protein levels were detected by ELISA. Tm increased PGE2-induced IL-6 release. **P < 0.01 PGE2v.s. PGE2 + Tm. n = 5. Cropped gels were run under the same experimental conditions.
To clarify the signal component responsible for this synergistic effect, we measured the influence of the PGE2 treatment alone on IL-6 levels under ER stress. Similar to the results obtained with PGE2 + IFNγ, PGE2-induced IL-6 production was synergistically enhanced under ER stress (Fig. 2C).
cAMP was involved in the enhancement of IL-6 with ER stress
Because PGE2 was involved in the synergistic effect on IL-6 production with ER stress, we next attempted to identify the signal component responsible for this effect. PGE2 receptors are known as G-protein-coupled receptors (GPCRs) and have been shown to affect intracellular cAMP levels22. Thus, we speculated that cAMP may be involved in the enhancement of IL-6 levels under ER stress. We treated glial cells with forskolin, which activates adenylate cyclase and analyzed IL-6 production in ER-stressed cells. Similar to the results obtained for PGE2 (Fig. 2C), forskolin synergistically enhanced IL-6 production under ER stress (Fig. 3A). This effect was further confirmed by analyzing dibutyryl cyclic AMP (dbcAMP), an analogue of cAMP (Fig. 3B). Simultaneous treatment with dbcAMP and IFNγ also had a synergistic effect on IL-6 production under ER stress (Fig. 3C). This effect was regulated transcriptionally, because the induction of IL-6 was observed at the mRNA level (Fig. 3D). Protein kinase A (PKA) is one of the major proteins activated through cAMP, therefore, we examined the possible involvement of PKA on IL-6 production. We pre-treated cells with H-89, a PKA inhibitor, for 30 min and then analyzed PGE2 + IFNγ-induced IL-6 production under ER stress. PGE2 + IFNγ-induced IL-6 production was markedly enhanced in cells treated with tunicamycin (an ER stress-inducing reagent). However, a significant reduction was not observed in IL-6 production in H-89 treated cells (Supplementary Fig S1). Thus, these results suggest that PKA may not be involved in the present stimuli. From these results, we can conclude that IL-6 production was mediated through cAMP, but not through PKA. Because the PKA-independent effect of cAMP has been implicated in the regulation of biological functions23, it would be interesting to examine these possibilities in future studies.

cAMP may be involved in the enhancement of IL-6 release under ER stress.
Glial cells were treated with tunicamycin (Tm: 1 μg/mL) for 1 h and then stimulated with (A) forskolin (10 μM), (B) dibutyryl cyclic AMP (dbcAMP: 1 mM), or (C) dibutyryl cyclic AMP (dbcAMP: 1 mM) + IFNγ (20 ng/mL) for 24 h. IL-6 protein levels were detected by ELISA. cAMP-induced IL-6 release was enhanced under ER stress. **P < 0.01, ***P < 0.001 forskolin, dbcAMP, dbcAMP + IFNγ v.s. Tm + forskolin or Tm + dbcAMP or Tm + dbcAMP + IFNγ. n = 4 ~ 9/group. (D) Glial cells were treated with tunicamycin (Tm: 1 μg/mL) for 1 h and then stimulated with dibutyryl cyclic AMP (dbcAMP: 1 mM) + IFNγ (20 ng/mL) for 4 h. IL-6 mRNA levels were detected by RT-PCR. Tm increased dbcAMP + IFNγ-induced IL-6 expression. **P < 0.01 dbcAMP + IFNγ v.s. dbcAMP + IFNγ + Tm. n = 6. The GAPDH was analyzed as loading control. Full-length gels are presented in Supplementary Fig S3. Full-length gels for GAPDH are presented in Supplementary Fig S4. Cropped gels were run under the same experimental conditions.
PGE2 + IFNγ-induced iNOS expression was inhibited under ER stress
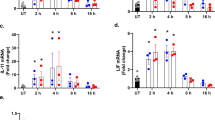
We next investigated the possible effect of ER stress on iNOS expression, which has been shown to increase under inflammatory conditions. Interestingly, in contrast to IL-6, we did not observe the induction of iNOS at the mRNA or protein level under ER stress (Figs. 4A and 4B). Furthermore, PGE2 + IFNγ-induced iNOS production was significantly inhibited, rather than increased (Fig. 4A). On the other hand, the expression of iNOS at the mRNA level was not inhibited under ER stress (Fig. 4B). Therefore, iNOS production appears to have been post-transcriptionally inhibited under ER stress, which was different from the regulatory mechanism of IL-6.

PGE2 + IFNγ-induced iNOS expression was inhibited at the protein level under ER stress.
(A) Glial cells were treated with tunicamycin (Tm: 1 μg/mL) for 1 h and then stimulated with PGE2 (15 μM) + IFNγ(20 ng/mL) for 24 h iNOS protein levels were detected by Western blotting. PGE2 + IFNγ-induced iNOS expression was inhibited under ER stress. ***p < 0.001 PGE2 + IFNγ v.s. PGE2 + IFNγ + Tm. n = 6. The GAPDH was analyzed as loading control. The intensities of the bands were expressed as iNOS/GAPDH. Full-length blots are presented in Supplementary Fig S5. (B) Glial cells were treated with tunicamycin (Tm: 1 μg/mL) for 1 h and then stimulated with PGE2 (15 μM) + IFNγ(20 ng/mL) for 4 h and RT-PCR analysis was performed. PGE2 + IFNγ-induced iNOS expression at the mRNA level was not inhibited under ER stress. n = 6. The GAPDH was analyzed as loading control. Full-length gels are presented in Supplementary Fig S4. Cropped gels/blots were run under the same experimental conditions. Between the cont and PGE2 + IFNγ conditions for figure 4B (iNOS), cropped blots have been presented together for display purposes.
cAMP was involved in the inhibition of iNOS under ER stress
We subsequently investigated the possible role of cAMP in down-regulating iNOS expression under ER stress. We treated glial cells with dbcAMP + IFNγ and analyzed iNOS levels. Similar to the results obtained with PGE2 + IFNγ (Fig. 4), we observed the significant inhibition of iNOS production under ER stress (Fig. 5A). On the other hand, dbcAMP + IFNγ-induced iNOS production at the mRNA level was not inhibited (Fig. 5B). Therefore, cAMP was involved in inhibiting iNOS through post-transcriptional mechanisms under ER stress.

cAMP may be involved in the inhibition of iNOS under ER stress.
(A) Glial cells were treated with tunicamycin (Tm: 1 μg/mL) for 1 h and then stimulated with dibutyryl cyclic AMP (dbcAMP: 1 mM) + IFNγ (20 ng/mL) for 24 h. iNOS protein levels were detected by Western blotting. dbcAMP + IFNγ-induced iNOS expression at the protein level was inhibited under ER stress. **p < 0.01 dbcAMP + IFNγ v.s. dbcAMP + IFNγ + Tm. n = 8. The GAPDH was analyzed as loading control. The intensities of the bands were expressed as iNOS/GAPDH. Full-length blots are presented in Supplementary Fig S6. (B) Glial cells were treated with tunicamycin (Tm: 1 μg/mL) for 1 h and then stimulated with dbcAMP (1 mM) + IFNγ (20 ng/mL) for 4 h. iNOS mRNA levels were detected by RT-PCR. dbcAMP + IFNγ-induced iNOS expression at the mRNA level was not inhibited under ER stress. n = 5. The GAPDH was analyzed as loading control. Full-length gels are presented in Supplementary Fig S4. Cropped gels/blots were run under the same experimental conditions.
Discussion
Inflammatory responses as well as ER stress are activated during neurodegenerative diseases. However, the link between these two responses remains unclear. In the present study, we showed unique mechanisms of association. We demonstrated that PGE2 + IFNγ-induced IL-6 expression was up-regulated, whereas iNOS expression was down-regulated in ER-stressed glial cells. ER stress has been shown to generally activate the PERK-eIF2α pathway to inhibit protein translation24,25. However, the molecular chaperon glucose regulated protein 78 (GRP78) and apoptotic transcription factor CHOP were shown to be up-regulated under these conditions26,27. Therefore, our observation of a synergistic effect on PGE2 + IFNγ-induced IL-6 production under ER stress is of particular interest. Therefore, we attempted to identify the mechanisms responsible for this effect. The receptor subtypes of PGE2 have been classified as EP1, EP2, EP3 and EP4. Astrocytes express the receptor for PGE228. These receptors are known as GPCRs and affect intracellular cAMP levels22. Therefore, we examined the possibility that cAMP may be involved in these processes. We showed the cAMP was involved in the synergistic induction of IL-6 under ER stress. We also demonstrated that treatment with ER stress alone caused a slight increase in IL-6 production (Fig. 2A). This effect may be mediated through XBP-1 because we detected an increase in XBP1 splicing in ER-stressed glial cells (Fig. 1) and the spliced form of XBP-1 was previously shown to induce IL-6 expression in B cells29. In contrast to IL-6, we observed the significant inhibition of PGE2 + IFNγ-induced iNOS expression in ER-stressed glial cells (Fig. 4A). This inhibitory effect of ER stress on iNOS expression may be mediated post-transcriptionally because we did not observe any inhibition at the mRNA level (Fig. 4B). The mechanisms responsible for these findings are at present unknown, but may be mediated through a translational inhibitory effect, such as the PERK-eIF2α pathway activated by ER stress (Fig. 1)24,25.
NO at low concentrations has been shown to be involved in multiple physiological functions such as neurotransmission and vasodilation30. However, excess amounts of NO, commonly produced by iNOS under inflammatory conditions, contribute to the development of vascular disease, diabetes and neurodegenerative disease. For example, excessive NO is produced from immune-activated pancreatic β cells in diabetes. Interestingly, this NO was reported to induce ER stress and cause apoptosis in pancreatic β cells31. Furthermore, NO was shown to inhibit protein-disulphide isomerase (PDI) activity by S-nitrosylation, which subsequently activated the UPR32. These findings led to the hypothesis that too much NO is toxic to cells and causes ER stress. On the other hand, NO was shown to protect against N-methyl-D-aspartate (NMDA)-mediated neurotoxicity in a physiological setting33. In the present study, we demonstrated that ER stress itself did not induce, but rather inhibited iNOS induced by PGE2 + IFNγ in glial cells (Fig. 4A). The present results raise questions regarding the physiological significance of these findings. Information on the stressed status of glial cells may be transmitted to neuronal cells, as neuron-glial communication plays an important role in maintaining normal brain functions34. Glial cells generally have both pro- and anti-apoptotic properties against neurons. In the present study, it is possible that glial cells may attenuate neuronal cell death associated with neurodegenerative diseases by inhibiting excessive NO production. Therefore, glial cells may be activating negative feedback regulatory signals to resist such stress. Additionally, the synergistic enhancement of IL-6 production from glial cells under ER stress (Fig. 2A) may contribute to neuroprotection because IL-6 has been shown to inhibit neuronal cell death35,36,37,38.
In the present study, we showed the up- and down-regulation of PGE2 + IFNγ-induced IL-6 and iNOS expression, respectively, under ER stress in glial cells. These results suggest that glial cells regulate immune function under ER stress. As ER stress is involved in neurodegenerative diseases such as AD, PD and cerebral ischemic insults, our results may uncover a new mechanism underlying neurodegenerative disorders and shed light on potential pharmacological treatments.
Methods
Materials
Tunicamycin (Tm) was obtained from Wako Pure Chemical Ltd. (Japan).
Preparation of primary cultured glial cells
Glial cells were prepared from the whole brains of neonatal C57BL/6 mice as described previously39. The cells were allowed to grow to confluency (10 days) in DMEM with 10% FCS, 100 units/ml penicillin G and 100 mg/ml streptomycin. All cultured cells were kept at 37°C in 5% CO2/95% air. Subsequently, mixed glial cells were shaken at 120 rpm for 18 h and cultured again for 4 to 6 days in 35-mm dishes. At this point, astrocyte cultures were routinely >95% positive for glial fibrillary acidic protein.
Gene expression analysis
Total RNA was isolated using TRI Reagent (Sigma-Aldrich, St. Louis, MO, USA). Reverse transcriptase-polymerase chain reaction (RT-PCR) was performed as described previously39. cDNA was synthesized from 2 μg of total RNA by reverse transcription using 25 U of Superscript Reverse Transcriptase(Invitrogen) and 0.25 μg of Oligo(dt)12–18 primer (Invitrogen) in a 20-μl reaction mixture containing First-Strand Buffer (Invitrogen), 1 mM dNTP mix, 10 mM DTT and 20 U of RNaseOUT Recombinant Ribonuclease Inhibitor (Invitrogen). Total RNA and the Oligo (dt) 12–18 primer were pre incubated at 70°C for 10 min prior to the reverse transcription. After incubation for 1.5 h at 46°C, the reaction was terminated by incubating samples for 15 min at 70°C. For PCR amplification, 1.2 μl of cDNA was added to 10.8 μl of a reaction mix containing 0.2 μM of each primer, 0.2 mM of dNTP mix, 0.6 U of Taq polymerase (Roche Diagnostics) and reaction buffer. PCR was performed in a DNA Thermal Cycler (MJ Research, PTC-220). The following primer sequences were used: IL-6; upstream, 5′–gtt ctc tgg gaa atc gtg ga-3′ and downstream, 5′-tgt act cca ggt agc tat gg-3′, iNOS; upstream, 5′–tca ctg gga cag cac aga at-3′ and downstream, 5′-tgt gtc tgc aga tgt gct ga-3′, GAPDH; upstream, 5′-aaa ccc atc acc atc ttc cag-3′ and downstream, 5′-agg ggc cat cca cag tct tct-3′. The PCR products (10 μl) were resolved by electrophoresis in an 8% polyacrylamide gel in TBE buffer. The gel was stained with ethidium bromide and photographed under ultraviolet light. The density of each band was measured using Image J 1.37v (Wayne Rasband, NIH) software. We confirmed that the increase in the intensity of the amplified product was increased according to the number of PCR cycles (Supplementary Fig S2).
Measurement of IL-6 levels
The cultured medium was centrifuged at 800 G at for 5 min 4°C and the supernatant was used as the sample. IL-6 levels were measured using a ELISA (DuoSet® ELISA Development kit (R&D Systems, USA)) according to the manufacturer's directions.
Western blotting
Cells were washed with ice-cold PBS and lysed in a buffer containing 10 mM HEPES-NaOH (pH 7.5), 150 mM NaCl, 1 mM EGTA, 1 mM Na3VO4, 10 mM NaF, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mM PMSF and 1% NP-40 for 20 min. The lysate was centrifuged at 15,000 rpm for 20 min at 4°C and the supernatant was collected. The samples were boiled with laemmli buffer for 3 min, fractionated by SDS-PAGE and transferred at 4°C to nitrocellulose membranes. The membranes were incubated with anti-KDEL (StressGen; diluted to 1:1,000), anti-CHOP (Santa Cruz; diluted to 1:500), anti-IRE1α (Cell Signaling; diluted to 1:1,000), anti-PERK (Cell Signaling; diluted to 1:1,000) and anti-XBP1 (Biolegend; diluted to 1:500) antibodies followed by anti-horseradish peroxidase-linked antibody. Peroxidase was detected by chemiluminescence using an ECL system.
Statistics
Results are expressed as the mean ± S.E. Statistical analyses were performed using the Student's t-test or paired t-test.
References
Sherman, M. Y. & Goldberg, A. L. Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron 29, 15–32 (2001).
Lindholm, D., Wootz, H. & Korhonen, L. ER stress and neurodegenerative diseases. Cell Death Differ 13, 385–392 (2006).
Hosoi, T. & Ozawa, K. Endoplasmic reticulum stress in disease: mechanisms and therapeutic opportunities. Clin Sci (Lond) 118, 19–29 (2010).
Hosoi, T. et al. TEK/Tie2 is a novel gene involved in endoplasmic reticulum stress. J Pharmacol Sci 114, 230–233 (2010).
Hosoi, T. & Ozawa, K. Molecular approaches to the treatment, prophylaxis and diagnosis of Alzheimer's disease: endoplasmic reticulum stress and immunological stress in pathogenesis of Alzheimer's disease. J Pharmacol Sci 118, 319–324 (2012).
Forman, M. S., Trojanowski, J. Q. & Lee, V. M. Neurodegenerative diseases: a decade of discoveries paves the way for therapeutic breakthroughs. Nat Med 10, 1055–1063 (2004).
Imai, Y. et al. An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell 105, 891–902 (2001).
Ryu, E. J. et al. Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson's disease. J Neurosci 22, 10690–10698 (2002).
Katayama, T. et al. Presenilin-1 mutations downregulate the signalling pathway of the unfolded-protein response. Nat Cell Biol 1, 479–485 (1999).
Hoozemans, J. J. et al. The unfolded protein response is activated in Alzheimer's disease. Acta Neuropathol 110, 165–172 (2005).
Hoozemans, J. J. et al. The unfolded protein response is activated in pretangle neurons in Alzheimer's disease hippocampus. Am J Pathol 174, 1241–1251 (2009).
Nakagawa, T. et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature 403, 98–103 (2000).
Hitomi, J. et al. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Aβ-induced cell death. J Cell Biol 165, 347–356 (2004).
Martinon, F., Chen, X., Lee, A. H. & Glimcher, L. H. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol 11, 411–418 (2010).
Woo, C. W. et al. Adaptive suppression of the ATF4-CHOP branch of the unfolded protein response by toll-like receptor signalling. Nat Cell Biol 11, 1473–1480 (2009).
Hosoi, T., Okuma, Y. & Nomura, Y. Electrical stimulation of afferent vagus nerve induces IL-1beta expression in the brain and activates HPA axis. Am J Physiol Regul Integr Comp Physiol 279, R141–147 (2000).
Hosoi, T., Okuma, Y. & Nomura, Y. The mechanisms of immune-to-brain communication in inflammation as a drug target. Curr Drug Targets Inflamm Allergy 1, 257–262 (2002).
Hopkins, S. J. & Rothwell, N. J. Cytokines and the nervous system. I: Expression and recognition. Trends Neurosci 18, 83–88 (1995).
O'Banion, M. K., Miller, J. C., Chang, J. W., Kaplan, M. D. & Coleman, P. D. Interleukin-1 beta induces prostaglandin G/H synthase-2 (cyclooxygenase-2) in primary murine astrocyte cultures. J Neurochem 66, 2532–2540 (1996).
Wyss-Coray, T. & Mucke, L. Inflammation in neurodegenerative disease--a double-edged sword. Neuron 35, 419–432 (2002).
Lucin, K. M. & Wyss-Coray, T. Immune activation in brain aging and neurodegeneration: too much or too little? Neuron 64, 110–122 (2009).
Sugimoto, Y. & Narumiya, S. Prostaglandin E receptors. J Biol Chem 282, 11613–11617 (2007).
Sands, W. A. & Palmer, T. M. Regulating gene transcription in response to cyclic AMP elevation. Cell Signal 20, 460–466 (2008).
Harding, H. P., Zhang, Y. & Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397, 271–274 (1999).
Shi, Y. et al. Identification and characterization of pancreatic eukaryotic initiation factor 2 α-subunit kinase, PEK, involved in translational control. Mol Cell Biol 18, 7499–7509 (1998).
Kozutsumi, Y., Segal, M., Normington, K., Gething, M. J. & Sambrook, J. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature 332, 462–464 (1988).
Harding, H. P. et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell 6, 1099–1108 (2000).
Fiebich, B. L., Schleicher, S., Spleiss, O., Czygan, M. & Hüll, M. Mechanisms of prostaglandin E2-induced interleukin-6 release in astrocytes: possible involvement of EP4-like receptors, p38 mitogen-activated protein kinase and protein kinase C. J Neurochem 79, 950–958 (2001).
Iwakoshi, N. N. et al. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat Immunol 4, 321–329 (2003).
Garthwaite, J. & Boulton, C. L. Nitric oxide signaling in the central nervous system. Annu Rev Physiol 57, 683–706 (1995).
Oyadomari, S. et al. Nitric oxide-induced apoptosis in pancreatic beta cells is mediated by the endoplasmic reticulum stress pathway. Proc Natl Acad Sci U S A 98, 10845–10850 (2001).
Uehara, T. et al. S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature 441, 513–517 (2006).
Lei, S. Z. et al. Effect of nitric oxide production on the redox modulatory site of the NMDA receptor-channel complex. Neuron 8, 1087–1099 (1992).
Fields, R. D. & Stevens-Graham, B. New insights into neuron-glia communication. Science 298, 556–562 (2002).
Carlson, N. G. et al. Inflammatory cytokines IL-1α, IL-1β, IL-6 and TNF-α impart neuroprotection to an excitotoxin through distinct pathways. J Immunol 163, 3963–3968 (1999).
Hirota, H., Kiyama, H., Kishimoto, T. & Taga, T. Accelerated Nerve Regeneration in Mice by upregulated expression of interleukin (IL) 6 and IL-6 receptor after trauma. J Exp Med 183, 2627–2634 (1996).
Loddick, S. A., Turnbull, A. V. & Rothwell, N. J. Cerebral interleukin-6 is neuroprotective during permanent focal cerebral ischemia in the rat. J Cereb Blood Flow Metab 18, 176–179 (1998).
Yamada, M. & Hatanaka, H. Interleukin-6 protects cultured rat hippocampal neurons against glutamate-induced cell death. Brain Res 643, 173–180 (1994).
Hosoi, T., Kume, A., Otani, K., Oba, T. & Ozawa, K. A unique modulator of endoplasmic reticulum stress-signalling pathways: the novel pharmacological properties of amiloride in glial cells. Br J Pharmacol 159, 428–437 (2010).
Acknowledgements
This research was supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan; Takeda Science Foundation; Tokyo Biochemical Research Foundation; Suzuken Memorial Foundation and The Nakajima Foundation.
Author information
Authors and Affiliations
Contributions
T.H. and K.O. designed research; T.H., M.H. and T.O. performed research; T.H., M.H., T.O. and K.O. analyzed data; and T.H. wrote the paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Hosoi, T., Honda, M., Oba, T. et al. ER stress upregulated PGE2/IFNγ-induced IL-6 expression and down-regulated iNOS expression in glial cells. Sci Rep 3, 3388 (2013). https://doi.org/10.1038/srep03388
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep03388
This article is cited by
-
Single-Cell Transcriptome Analysis Reveals the Role of Pancreatic Secretome in COVID-19 Associated Multi-organ Dysfunctions
Interdisciplinary Sciences: Computational Life Sciences (2022)
-
ER stress and genomic instability induced by gamma radiation in mice primary cultured glial cells
Metabolic Brain Disease (2018)
-
Endoplasmic reticulum stress and inflammation in the central nervous system
Molecular Neurodegeneration (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
