
Abstract
Identifying breast cancer patients is crucial to the clinical diagnosis and therapy for this disease. Conventional gene-based methods for breast cancer diagnosis ignore gene-gene interactions and thus may lead to loss of power. In this study, we proposed a novel method to select classification features, called “Selection of Significant Expression-Correlation Differential Motifs” (SSECDM). This method applied a network motif-based approach, combining a human signaling network and high-throughput gene expression data to distinguish breast cancer samples from normal samples. Our method has higher classification performance and better classification accuracy stability than the mutual information (MI) method or the individual gene sets method. It may become a useful tool for identifying and treating patients with breast cancer and other cancers, thus contributing to clinical diagnosis and therapy for these diseases.
Similar content being viewed by others

Introduction
Breast cancer is a heterogeneous disease and identification of this disease is a major clinical challenge. The recovery rate of patients diagnosed in the earliest stages of breast cancer approaches 95%1. Genome-wide high-throughput expression data provide a valuable platform to identify disease markers for breast cancer2,3. However, data for individual gene do not discover the molecular mechanisms responsible for these determinations4 and individual signatures are less reproducible in different breast cancer groups5. In contrast, network-based methods for classification have been shown to be more reproducible than methods based on individual genes6. However, existing available methods cannot determine whether signal transduction was systematically disturbed in tumor cells. Disruption of the signaling network might trigger key signals such as cell proliferation or evading growth suppressors for uncontrolled growth and promoting tumor progression; it might also inhibit tumor-suppressors, leading to an imbalance between cell growth and apoptosis7.Changes in the signaling network not only indicate disruptions leading to carcinogenesis, but also reveal the changes in expression-correlated differential between normal and tumor conditions8. Therefore, a better approach to classifying breast cancer samples may be exploiting datasets covering both the human signaling network and breast cancer gene expression profiles.
Mutual information is widely used as a generalized correlation measure9. Cicek AE et al. proposed a new multivariate technique (ADEMA) based on MI to identify expected metabolite level changes with respect to a specific condition and showed that ADEMA predicts De Novo Lipogenesis pathway metabolite level changes in samples with Cystic Fibrosis (CF) better than the prediction method based on the significance of individual metabolite level changes. ADEMA results had up to 31% higher accuracy as compared to other classification algorithms10. Network motifs are small, repeated and conserved biological units ranging from molecular domains to small reaction networks and thus serve as building blocks of network structures6,11. Lizier JT et al. investigated the role of two- and three-node motifs in contributing to local information storage12. Choi J et al. adopted the Typed Network Motif Comparison Algorithm (TNMCA) to infer novel drug indications using topology of given network13.Shellman ER et al. presented a comparative analysis of motif distributions in the metabolic networks of 21 species across six kingdoms of life14. Wu SF et al. explored the mechanisms of cervical carcinoma response to epidermal growth factor (EGF) using network motifs in the regulation network15. Yuji Zhang et al. presented a novel network motif-based approach that integrates biological network topology and high-throughput gene expression data to identify markers not as individual genes but as network motifs. To determine significant network motifs, researchers calculated their activity score based on MI using gene expression data, which was more reproducible than individual gene markers selected without network information6. The MI method requires standardization and discretization of expression data in the calculation of activity score, which may reduce the authenticity of the data. It guarantees the authenticity of data to start directly from the original expression data and assess significance of motifs using the expression correlation among genes.
Here, we propose a network motif-based method for selecting high-stability significant expression-correlation differential motifs (HSCDMs) to classify breast cancer samples by integrating the human signaling network and gene expression profiles. SSECDM method could potentially be applied to the identification of breast cancer patients using unknown samples.
Results
Network motifs
We applied Cytoscape16 to analyze the global properties of the human signaling network. Cytoscape is a software environment that is used to display, analyze and edit graphical network. With network data imported into Cytoscape and the network analysis plugin selected, Cytoscape can comprehensively analyze the signaling network. The signaling network followed a power law degree distribution and displayed the small world property. Genes were labeled if they were cancer genes in the network. It was found that the average degrees of the whole network, the cancer genes and the breast cancer genes were 6.3, 9.97 and 23.44, respectively. These results suggest that cancer genes, especially breast cancer genes, have pivotal positions in the signaling network (Fisher's exact test, p = 0.002 for the genes of the whole network and the breast cancer genes and p = 0.041 for the cancer genes and the breast cancer genes, respectively).
To explore the role of cancer-associated motifs in the signaling network, we screened the basic skeleton of three-node motifs17. We found 11,150 significant network motifs (p < 0.05) that appeared at least five for 1000 random disturbances on the human signaling network using FANMOD18. Of them, 3549 motifs were associated with cancer.
HSCDMs
Using four expression profiles (GSE5364, GSE9574, GSE15852, GSE20437, the same platform GPL96) with disease and normal samples downloaded from the GEO database (Table 1), we first calculated expression-correlation differential scores for each motif and then plotted them against the normal distribution (Figure 1).

Normal distribution plotted against expression correlation differential scores for breast cancer expression profile dataset GSE5364.
X-axis: expression-correlation differential score for network motifs. Y-axis: number of network motifs. Red arrow: score at which p-value equals 0.05.
To improve the stability of our method, we selected the significant expression-correlation differential motifs (SCDMs) that occurred in at least two datasets and defined them as HSCDMs (Table 2). Fifty-six HSCDMs (labeled M1 to M56) were found in the four expression profile datasets. It was shown that, 51 of the identified HSCDMs (91%) were associated with breast cancer19,20,21,22,23(Supplementary table 1).
M1 occurred in four datasets and M2 occurred in three datasets, M3–M56 occurred only in two datasets. 81.18% (69/85) of the genes in 56 HSCDMs were associated with breast cancer. All of the three genes of each motif in M1–M35 and two genes of any motif in M36–M51 are highly associated with breast cancer. For example, BCL2, BINP3 and MAP2K1 in M1 were reported in literature (Figure 2). BCL2 is a known cancer gene of chronic lymphocytic leukemia and is a highly prognostic factor for breast cancer24. BINP3 protect cells from virus-induced cell death and is a prognostic marker in breast cancer25, as well as an anti-cancer target26. MAP2K1 (known as MEK1) integrates many biochemical signals and is involved in multiple cellular processes, including proliferation, development and differentiation. Suppression of MEK1 blocked MED28-induced MMP2 activation, cellular migration and invasion in breast cancer cells. Moreover, MED28 is highly expressed in breast cancer, overexpression of which enhanced cellular migration and up-regulated MMP2 and MEK1 expression27.Three genes in M2 (AKT1, HSP90 and GEF) were reported to be related with breast cancer (Figure 2). Recurrent somatic mutations in AKT1 are found in breast cancer28. HSP90 is an important anti-cancer target and its inhibition can induce cell cycle arrest and apoptosis in human breast cancer cells29. GEF can induce apoptosis in MCF-7 breast cancer cells and is also a breast cancer target gene in clinical therapy30. CBP was found in M10, M11, M16, M22, M31 and M33. CBP/p300-interacting transactivator with Glu/Asp-rich carboxy-terminal domain 2 (CITED2) is a member of the CITED family and is exclusively recruited to the promoter region of the FVII gene specifically in breast cancer cells31. CITED2 was recently verified to be over expressed in human breast tumors relative to normal mammary epithelium32. c-JUN, the first cellular overexpressed proto-oncogene in human breast cancer33, was found in M10, M11 and M22. The activation of CTBP1 (C-terminal binding protein 1), which was found in M11, could be a potential biomarker for breast cancer development since CTBP1 could down-regulated Brca1 and E-cadherin in breast cancer34. CREB (found in M16, M37, M41, M45–M48, M51 and M54) played an important role in antimigratory in breast cancer cells35. We note that our cancer-associated motifs were based on all types of cancer genes. It could be believed that mapping breast cancer expression profile datasets to HSCDMs, we could obtain highly associated genes of breast cancer.

High stable significant differential motifs.
Solid lines represent activating or inhibitory interactions. Dotted lines represent physical interactions. Nodes in pink or green represent cancer-associated or non-cancer-associated genes, respectively.
Therefore, the identified HSCDMs were demonstrated to be associated with breast cancer. In contrast, traditional differential expression analysis did not identify the majority of the genes in the identified HSCDMs (Table 3).
To investigate the importance of HSCDMs in terms of topological properties of the network, each HSCDM was individually removed from the signaling network. For each derived network, we then calculated the average number of neighbors and the characteristic path length. Next, each random non-HSCDMs were individually removed from the signaling network to generate reference networks and the calculations were repeated. This randomization process was performed 100 times, generating 100 randomized networks. The average number of neighbors was significantly lower when HSCDMs were removed than when random non-HSCDMs were removed and the characteristic path length was significantly longer when HSCDMs were removed than when random non-HSCDMs were removed (Figure 3, permutation test, P < 0.05). These results imply that HSCDMs play a prominent biological role in the signaling network.

Network topology characteristics for high-stability significant differential motifs.
(A) Average number of neighbors. (B) Characteristic path length. Red dots: Average number of neighbors and characteristic path length of original signaling network,signaling network with motif M1 removed, signaling network with motif M2 removed (2GSE). Box plots summarize results from 100 random networks.
Classification of HSCDMs
Classification accuracy was estimated by using leave-one-out cross validation (LOOCV), with the average expression level of genes in the 56 identified HSCDMs as classification features and a Support Vector Machine (SVM) as the classifier. The classification performances of four kernel functions (linear, quadratic, polynomial and radial basis kernel functions) were evaluated. The classification accuracy was highest and most stable when the linear kernel function was used (Table 4). Therefore, we chose linear kernel function in our subsequent analyses.
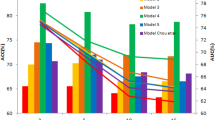
Because the number of samples might influence classification accuracy, the dataset with the largest number of samples (GSE5364) was divided into proportional gradients and each defined proportion was compared to 100 randomized selections with the same number of samples (Figure 4). It showed that the sample gradient increased, the classification accuracy increased. Even in small gradient, classification accuracy of our method was higher than 85%. In Table 5, the classification accuracies for the other three expression profilings with small sample sizes were also higher than 80%. As we know, in the case of large samples, any method could improve the classification accuracy. Our method not only confirmed this, but also had high classification accuracies among the small sample sizes. These results suggested that our method had good stability of classification accuracy.

The influence of different sample gradients on classification accuracy.
X-axis: the proportion of normal samples to tumor samples. Y-axis: classification accuracy using the given normal-to-tumor sample proportion. Box plot summarizes 100 randomized selections of normal and tumor samples.
Classification evaluation based on individual gene sets
We compared the classification accuracy of HSCDMs to the classification accuracy of individual gene sets, i.e. HSCDMs' genes, breast cancer genes, breast cancer marker genes (70 marker genes in Materials) and a set containing both breast cancer genes and breast cancer marker genes (Table 5). In Table 5, the classification accuracy of our classification feature in GSE5364 dataset (large sample), was slightly higher than ones of other classification features. However for the other three expression profilings (small sample, GSE9574, GSE15852, GSE20437), the classification accuracy of our method was almost higher than ones of all individual gene approaches. At the same time for expression profilings of small sample size, the classification accuracy of the HSCDMs' genes as classification feature was slightly higher than ones with the other individual gene classification features. It suggested the HSCDMs' genes may be highly involved in development of breast cancer, which was confirmed through literature reviewed that 81% (69/85) of the HSCDMs' genes were association with breast cancer. These results indicated that HSCDMs had not only a higher classification accuracy, but also higher classification accuracy stability. To further demonstrate the classification accuracy stability of our method, we chose GSE27562 (Table 1) on another platform (GPL570) and classified it using different classification features, the classification accuracy of different classification features were displayed in the last column of Table 5.
Classification evaluation based on mutual information
To further assess the performance of HSCDMs, we compared our results with those of another method based on mutual information for screening significant differential motifs36,6. The MI method applies an activity score to select classification features. First, as with our method, we identified network motifs within the human signaling network using FANMOD (p < 0.05, number of occurrences five or greater). Second, we calculated the activity score of each motif. Fitting a distribution of these scores, we defined significant mutual information differential motifs (SMIDMs) (permutation test, p < 0.05). To improve the stability of this method, we generated four groups of SMIDMs from four datasets (GSE5364, GSE9574, GSE15852 and GSE20437). SMIDMs occurring in at least two datasets were defined as high-stability significant mutual information differential motifs (HSMIDMs). We consider HSMIDMs to be classification features for positively identifying breast cancer samples. We found 6 HSMIDMs occurring in three datasets and 73 HSMIDMs occurring in two datasets, respectively. Third, we used these 79 HSMIDMs and the 56 HSCDMs found by SSECDM method as features for classifying the four breast cancer sample sets. ROC curves, which reflect the sensitivity and specificity for the two methods showed that the MI method AUC score was lower than the SM score (MI_AUC = 0.7567, SM_AUC = 0.8125). These results suggested that our approach had superior classification performance.
Discussion
We proposed a network motif-based method for selecting classification features for the patients with breast cancer that integrates human signaling network and gene expression profiles. In this study, we compared the expression-correlation difference between normal and tumor conditions, using SCDMs generated from the signaling network. By screening SCDMs from multiple breast cancer datasets, HSCDMs were identified. HSCDMs were validated as cancer-associated motifs and individual genes from the HSCDMs were also validated as susceptible genes or prognostic factors for breast cancer by reviewing the recent literatures. Compared with the naive Bayes classifier, the SVM classifier had better classification performance using 56 HSCDMs as classification features (Table 6). Naive Bayesian classifier is based on a simple assumption that attributes are conditional independent when target value was given. The assumption is often not met in practical applications, which influences the correct classification of Naive Bayesian classifier. However, SVM classifier could optimize itself and output the optimized results. It was found that the majority of genes in HSCDMs were not identified by differential expression analysis alone (Table 3). SSECDM method was then compared to MI and the method based on individual genes. It was shown that SSECDM method for classifying breast cancer samples using the four datasets had the highest accuracy for predicting breast cancer.
Furthermore, our results suggest that HSCDMs can potentially serve as prognostic markers of breast cancer. Assuming that a larger expression correlation differential score for a motif, implies a shorter survival time, this score could predict the survival time of breast cancer patients. To test this hypothesis, we obtained three datasets of breast cancer expression profiles with survival information (GSE1456, GSE3494 and GSE6532) and analyzed the relationship between survival time and HSCDMs. We found that 15 of the identified HSCDMs (26.8%) were significantly negatively correlated with the survival time of breast cancer patients (t-test, p < 0.05), suggesting that these motifs with negative correlation could be promising potential prognostic markers of breast cancer. These motifs with negative correlation could add value to clinical application.
In conclusion, our method is capable of improving the accuracy of identifying patients with breast cancer and may aid in the clinical diagnosis and therapy of breast cancer and other types of cancer.
Methods
We proposed a novel method to select classification features, called “Selection of Significant Expression-Correlation Differential Motifs”. SSECDM method applied a network motif-based approach for selecting high-stability significant expression-correlation differential motifs to classify breast cancer samples by integrating the human signaling network and gene expression profiles (Figure 5). We further describe each step below.

The flowchart of SSECDM method.
μ indicates the average expression level of genes in HSCDM.
Materials
A human signaling network was obtained from a previous study, which contains 5089 interactions among 1634 genes37. The three types of interactions addressed are activation, inhibition and physical interaction.
A list of 458 cancer-associated genes was obtained from the Cancer Gene Census database38 (Downloaded in 2011). A list of 70 marker genes for breast cancer were obtained from a previous study39,6.
To integrate the gene expression and signal network datasets, we mapped the gene expression value of each gene onto the network. Gene expression datasets were extracted from the GEO database40 (Table 1).
Selection of classification features
The classification features were selected by screening for significant changes in gene expression between normal and tumor conditions. First, we extracted network motifs based on the human signaling network. Second, significant expression-correlation differential motifs were identified by comparing the network motif expression-correlation differential score for normal and tumor conditions using the gene expression datasets. Finally, to improve the method's stability, we obtained four groups of SCDMs from the four datasets. SCDMs that were found in at least two datasets were called high-stability significant expression-correlation differential motifs. We consider the average expression level of genes in HSCDMs to be classification features for positively identifying breast cancer samples.
Cancer-associated motifs
We applied FANMOD18 to select network motifs. A network motif was defined based on the criterion that the number of occurrences must be at least five and also must be significantly higher than that used in randomized networks. The significance test was carried out on 1000 randomized networks and a pattern with p < 0.05 was considered statistically significant.
Furthermore, we mapped the cancer genes to network motifs and defined the cancer-associated motifs if the network motifs contained at least of one cancer genes.
HSCDMs
Given a particular motif M with E1, E2 and E3 representing the three edges of M, the expression-correlation differential score S was defined:



Where (X, Y) and (X′, Y′) are the gene expression values under normal and tumor conditions, respectively. Ek and Ek' are the Pearson Correlation Coefficients of the kth edge connecting two genes under normal and tumor conditions, respectively.
Based on the expression-correlation differential score of each motif, we simulated the distribution of these scores and then defined SCDMs (permutation test, p < 0.05).
We identified SCDMs that occurred in at least two datasets and defined those as HSCDMs. We then used the average expression level of genes in HSCDMs as classification features to identify patients with breast cancer.
Classification and evaluation
We applied the SVM method, using four different kernel functions (linear, quadratic, polynomial and radial basis kernel) to classify patients with breast cancer. We then applied LOOCV to estimate the performance of our classification.
References
Vicini, F. et al. Differences in disease presentation, management techniques, treatment outcome and toxicities in African-American women with early stage breast cancer treated with breast-conserving therapy. Cancer 116, 3485–3492 (2010).
van de Vijver, M. J. et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 347, 1999–2009 (2002).
Duan, K. B., Rajapakse, J. C., Wang, H. & Azuaje, F. Multiple SVM-RFE for gene selection in cancer classification with expression data. IEEE Trans Nanobioscience. 4, 228–234 (2005).
Han-Yu, Eunjung, Yu-Tsueng, Doheon & Trey Network-based classification of breast cancer metastasis. BMC Syst Biol. 3, 140 (2007).
Ein-Dor, L., Zuk, O. & Domany, E. Thousands of samples are needed to generate a robust gene list for predicting outcome in cancer. Proc Natl Acad Sci U S A. 103, 5923–5928 (2006).
Zhang, Y., Xuan, J., de Los Reyes, B. G., Clarke, R. & Ressom, H. W. Network motif-based identification of breast cancer susceptibility genes. Conf Proc IEEE Eng Med Biol Soc 2008, 5696–5699 (2008).
Hanahan, D. & Weinberg, R. A. Hallmarks of Cancer: The Next Generation. Cell. 144, 646–674 (2011).
Schramm, G., Kannabiran, N. & König, R. Regulation patterns in signaling networks of cancer. BMC Syst Biol. 4, 162 (2010).
Song, L., Langfelder, P. & Horvath, S. Comparison of co-expression measures: mutual information, correlation and model based indices. BMC Bioinformatics 13, 328 (2012).
Cicek, A. E., Bederman, I., Henderson, L., Drumm, M. L. & Ozsoyoglu, G. ADEMA: an algorithm to determine expected metabolite level alterations using mutual information. PLoS Comput Biol 9, e1002859 (2013).
Alon, U. Network motifs: theory and experimental approaches. Nat Rev Genet 8, 450–461 (2007).
Lizier, J. T., Atay, F. M. & Jost, J. Information storage, loop motifs and clustered structure in complex networks. Phys Rev E Stat Nonlin Soft Matter Phys 86, 026110 (2012).
Choi, J., Kim, K., Song, M. & Lee, D. Generation and application of drug indication inference models using typed network motif comparison analysis. BMC Med Inform Decis Mak 13 Suppl 1, S2 (2013).
Shellman, E. R., Burant, C. F. & Schnell, S. Network motifs provide signatures that characterize metabolism. Mol Biosyst 9, 352–360 (2013).
Wu, S. F. et al. Network motifs in the transcriptional regulation network of cervical carcinoma cells respond to EGF. Arch Gynecol Obstet 287, 771–777 (2013).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504 (2003).
Milo, R. et al. Network motifs: simple building blocks of complex networks. Science 298, 824–827 (2002).
Wernicke, S. & Rasche, F. FANMOD: a tool for fast network motif detection. Bioinformatics. 22, 1152–1153 (2006).
Arnedos, M. et al. Array CGH and PIK3CA/AKT1 mutations to drive patients to specific targeted agents: a clinical experience in 108 patients with metastatic breast cancer. Eur J Cancer 48, 2293–2299 (2012).
Iacovides, D. C. et al. Identification and quantification of AKT isoforms and phosphoforms in breast cancer using a novel nanofluidic immunoassay. Mol Cell Proteomics (2013).
Thomas, S., Thurn, K. T., Raha, P., Chen, S. & Munster, P. N. Efficacy of histone deacetylase and estrogen receptor inhibition in breast cancer cells due to concerted down regulation of Akt. PLoS One 8, e68973 (2013).
Uji, K. et al. Significance of TP53 mutations determined by next-generation "deep" sequencing in prognosis of estrogen receptor-positive breast cancer. Cancer Lett S0304–3835, 00603–00604 (2013).
Youssef, G., Gillett, C., Agbaje, O., Crompton, T. & Montano, X. Phosphorylation of NTRK1 at Y674/Y675 induced by TP53-dependent repression of PTPN6 expression: A potential novel prognostic marker for breast cancer. Mod Pathol (2013).
Hwang, K. T. et al. Prognostic influence of BCL2 expression in breast cancer. Int J Cancer 131, E1109–E1119 (2012).
Naushad, S. M., Prayaga, A., Digumarti, R. R., Gottumukkala, S. R. & Kutala, V. K. Bcl-2/adenovirus E1B 19 kDa-interacting protein 3 (BNIP3) expression is epigenetically regulated by one-carbon metabolism in invasive duct cell carcinoma of breast. Mol Cell Biochem. 361, 189–195 (2012).
Knutson, A. K. et al. Comparative effects of histone deacetylase inhibitors on p53 target gene expression, cell cycle and apoptosis in MCF-7 breast cancer cells. Oncol Rep 27, 849–853 (2012).
Huang, C. Y., Chou, Y. H., Hsieh, N. T., Chen, H. H. & Lee, M. F. MED28 regulates MEK1-dependent cellular migration in human breast cancer cells. J Cell Physiol 227, 3820–3827 (2012).
Banerji, S. et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature 486, 405–409 (2012).
Wang, H. C. et al. Withanolides-induced breast cancer cell death is correlated with their ability to inhibit heat protein 90. PLoS One. 7, e37764 (2012).
Boulaiz, H. et al. gef Gene Expression in MCF-7 Breast Cancer Cells is Associated with a Better Prognosis and Induction of Apoptosis by p53-Mediated Signaling Pathway. Int J Mol Sci. 12, 7445–7458 (2011).
Koizume, S. et al. Hepatocyte nuclear factor-4-independent synthesis of coagulation factor VII in breast cancer cells and its inhibition by targeting selective histone acetyltransferases. Mol Cancer Res 7, 1928–1936 (2009).
Lau, W. M., Doucet, M., Huang, D., Weber, K. L. & Kominsky, S. L. CITED2 modulates estrogen receptor transcriptional activity in breast cancer cells. Biochem Biophys Res Commun 437, 261–266 (2013).
Jiao, X. et al. c-Jun induces mammary epithelial cellular invasion and breast cancer stem cell expansion. J Biol Chem 285, 8218–8226 (2010).
Deng, Y. et al. Transcriptional down-regulation of Brca1 and E-cadherin by CtBP1 in breast cancer. Mol Carcinog 51, 500–507 (2012).
Hansen, C. et al. Wnt-5a-induced phosphorylation of DARPP-32 inhibits breast cancer cell migration in a CREB-dependent manner. J Biol Chem 284, 27533–27543 (2009).
Tomlins, S. A. et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 310, 644–648 (2005).
Cui, Q. et al. A map of human cancer signaling. Mol Syst Biol 3, 152 (2007).
Futreal, P. A. et al. A census of human cancer genes. Nat Rev Cancer 4, 177–183 (2004).
van 't Veer, L. J. et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 415, 530–536 (2002).
Edgar, R., Domrachev, M. & Lash, A. E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30, 207–210 (2002).
Acknowledgements
This work was supported in part by the Science & Technology Research Project of the Heilongjiang Ministry of Education (Grant No. 12511271), the National Natural Science Foundation of China (Grant No. 61272388) and the Natural Science Foundation of Heilongjiang Province (Grant No. F201237).
Author information
Authors and Affiliations
Contributions
L.C. developed the theory, X.Q. and M.C. designed and supervised the experiments, Y.Z. and W.L. provided ideas and materials, W.L. wrote programs to process data, C.F. and X.J. performed the survival analysis, Y.H. and W.H. designed and manipulated figures 1–5, Y.Z. designed tables 1–6, B.L. reviewed and modified contents of the manuscript, L.C., X.Q. and M.C. wrote the manuscript. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Chen, L., Qu, X., Cao, M. et al. Identification of breast cancer patients based on human signaling network motifs. Sci Rep 3, 3368 (2013). https://doi.org/10.1038/srep03368
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep03368
This article is cited by
-
Hypergraph motifs and their extensions beyond binary
The VLDB Journal (2024)
-
Exact and sampling methods for mining higher-order motifs in large hypergraphs
Computing (2024)
-
Higher-order motif analysis in hypergraphs
Communications Physics (2022)
-
Transcription factor and microRNA-regulated network motifs for cancer and signal transduction networks
BMC Systems Biology (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
