
Abstract
Interleukin (IL)-32 is known to exert adujvant effects on innate immune response, however, receptors and downstream signaling pathways remain to be clarified. Here we found that IL-32γ upregulated serine protease activity of proteinase-3 (PR3), in turn triggering protease-activated receptor 2 (PAR2) signaling. Interestingly, silencing of PR3 or PAR2 using siRNA markedly diminished IL-32γ-induced TNFα and IFN-β mRNA expression. IL-32γ-PAR2 axis utilized TRIF and Ras-Raf-1 pathways. On stimulation with lipopolysaccharide (LPS), differential activation of protein kinase C isoforms modulated the balance between LPS-TLR4-TRIF and IL-32-PAR2-TRIF axes, because LPS was a strong inducer of IL-32γ. IL-32-PAR2-TRIF axis might serve not only as an extracellular sensor of bacterial and autologous proteases, but also as a modulator of innate and adaptive immunity during infection.
Similar content being viewed by others


Introduction
The peptide originally identified as natural killer (NK) transcript 41 was renamed as interleukin (IL)-32 after being shown to possess proinflammatory characteristics2. As research progressed, several isoforms of IL-32 were described. Initially, four isoforms (IL-32α, β, γ and δ) were found to derive from alternative splicing of a single gene. Among these, IL-32γ is the longest and offers the strongest biological activity2,3. Two additional isoforms have recently been identified, IL-32ε and ζ, but these isoforms are not ubiquitously expressed except in T cells4. Given that IL-32 has been identified only in higher mammals, in which adaptive immunity is highly developed2, it is tempting to speculate that the role of IL-32 is relevant to adaptive immunity itself or the transition from innate to adaptive immunity in the context of bacterial or virus infection.
IL-32 has been shown to induce various inflammatory cytokines, such as tumor necrosis factor (TNF)α, IL-1, IL-6 and IL-8. Due to such proinflammatory properties, IL-32 has been considered to play a key role in innate immunity host-defense and the development of chronic inflammatory diseases, including mycobacterial5,6 or viral infection7,8,9, rheumatoid arthritis (RA), inflammatory bowel disease (IBD)10 and chronic obstetric pulmonary disease11. Several publications have recently reported that IL-32 exerts a host defensive role, particularly against viral infections. Patients infected with human immunodeficiency virus (HIV) show elevated IL-32 serum levels and silencing endogenous IL-32 increases HIV p24 production8,12. Moreover, blockade of IFN-α/IFN-β bioactivity in IL-32γ-stimulated U1 macrophages results in enhanced production of HIV, demonstrating that the antiviral activity of IL-32γ is applied through these type I interferons (IFNs)12.
Mounting evidence regarding upstream signaling regulators for IL-32 production has been accumulating in the literature13,14,15,16,17. However, signaling pathways downstream of IL-32 have yet to be fully elucidated. We have previously shown that IL-32-induced TNFα production is mediated through nuclear factor (NF)-κB and extracellular signal-regulated protein kinase (ERK)1/2 mitogen-activated protein kinase (MAPK) in RAW264.7 cells18, but details of the IL-32 signaling cascade to ERK1/2 and NFκB activation have yet to be elucidated. Synergistic interactions of IL-32 with ligands of pattern recognition receptors (PRRs) such as nucleotide oligomerization domain (NOD)1/2 and toll-like receptor (TLR)2/4 have been described in a number of reports18,19,20, whereas IL-32 can substantially trigger the TLR signaling cascade without any PRR ligand18,20. Protease-activated receptor 2 (PAR2) is a potential candidate molecule capable of explaining IL-32 bioactivity including use of TLR signaling and type I IFN-mediated antiviral immunity. PAR2 has been shown to induce TNFα and type I IFN, predominantly through a myeloid differentiation factor 88 (MyD88)-independent TLR signaling pathway, that is a Toll/IL-1 receptor (TIR)-domain-containing adapter-inducing IFN-β (TRIF) signaling pathway, which is reportedly implicated in delayed kinetics of TLR4-mediated NF-κB activation.
PAR2 is a seven-transmembrane G protein-coupled receptor and acts as a PRR, sensing not only bacterial serine proteases, but also autologous serine proteinase, proteinase-3 (PR3) in the context of inflammation and infection. PR3 reportedly binds to IL-3221 and is capable of stimulating PAR222,23,24. Since membrane-bound PR3 expression increases in chronic inflammatory diseases such as vasculitis and RA25, activation of PAR2 by PR3 may occur on the cell surface. Among six isoforms of IL-32, IL-32γ is the isoform most likely to have the ability to be bound to membrane-bound PR3 and subsequently activate PAR2. The present study examined the extracellular biological function of IL-32γ through interactions with PR3 and PAR2, which ultimately resulted in triggering PAR2-TRIF signaling axis and proposes a potential role of IL-32 in the transition from innate to adaptive immunity.
Results
Lipopolysaccharide (LPS) is the strongest inducer of IL-32γ among multiple pathogen-associated molecular patterns (PAMPs) recognized by TLRs
While unstimulated THP-1 cells did not constitutively express IL-32 mRNA, either LPS- or zymosan-stimulated THP-1 cells expressed significantly high levels of IL-32 mRNA and IL-32 protein (Fig. 1a). On the other hand, little or no expression of IL-32 mRNA was apparent when THP-1 cells were stimulated with other TLR ligands, such as poly (I:C), imquimod and CpGDNA. The expression level of IL-32 and the amount of IL-32 protein induced by LPS was increasing, at least over the first 24 h following LPS-stimulation (Fig. 1b).

LPS is the strongest inducer of IL-32 among multiple PAMPs.
THP-1 cells were cultured with LPS (100 ng/ml or 1 μg/ml), zymosan (10 or 100 μg/ml), poly (I:C) (10 or 100 μg/ml), imiquimod (10 or 100 μg/ml) and CpG-B DNA (10 or 100 μM) for 6 h, then levels of IL-32 mRNA and protein in culture supernatant were measured by real-time PCR or specific ELISA. LPS and zymosan significantly induced IL-32 at both mRNA and protein levels, whereas other ligands did not (a). Temporal changes in the level of IL-32 induced by LPS (100 ng/ml) were measured at both mRNA and protein levels by real-time PCR or ELISA (b). The expression level of IL-32 mRNA and protein induced by LPS was increasing at least over the first 24 h following LPS stimulation. Values are expressed as mean ±SD for four independent experiments. *p < 0.05 and **p < 0.01.
Both IL-32γ and LPS can solely induce TNFα and IFN-β mRNA expression
Both IL-32γ and LPS were capable of independently stimulating TNFα mRNA expression and protein production by THP-1 cells, with much higher levels for LPS than for IL-32γ. Interestingly, LPS-induced TNFα mRNA expression and protein production demonstrated double peaks during the course of 48 h stimulation, whereas IL-32γ-induced TNFα mRNA expression and protein production displayed a single peak (Fig. 2a). LPS and IL-32γ also stimulated THP-1 cells to express IFN-β mRNA and produce IFN-β protein with comparable levels and a bimodal expression or production pattern was again observed particularly for LPS-stimulation (Fig. 2b). When THP-1 cells were transfected with siRNA targeting IL-32γ, double peaks of LPS-induced TNFα expression and production changed to a single peak, while each control did not show such effect (Fig. 2c), suggesting that the second peak observed at 12 h was attributable to the effects of LPS-induced IL-32γ. As for IFN-β, the first peak was observed at 6 h after IL-32γ stimulation; thereafter, the expression and production level was constant without an apparent second peak until 48 h, suggesting that the second peak of IFN-β is attributable to LPS-induced IL-32γ (Fig. 2d).

Both IL-32γ and LPS can independently induce TNFα and IFN-β mRNA expression and protein production.
LPS (100 ng) or IL-32γ (100 ng) was added to THP-1 cells and mRNA and protein levels of TNFα and IFN-β were measured by real-time PCR and ELISA. LPS induced both TNFα and IFN-β at mRNA and protein levels with an apparent bimodal pattern (a, b). While IL-32γ could also induce both TNFα and IFN-β at mRNA and protein levels, the pattern was monomodal (a, b). RNA interference with siRNA (si) targeting IL-32 resulted in a monomodal pattern of TNFα and IFN-β expression induced by LPS while each negative control (ct-si) did not exhibit this effect (c, d). Values are expressed as mean ±SD for four independent experiments.
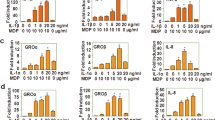
Activation of PR3 is indispensable for IL-32γ-induced TNFα and IFN-β mRNA expression
Although PR3, a serine protease, reportedly acts as a binding protein for IL-3223, whether IL-32γ modulates the serine protease activity of PR3 remains unclear. PR3 activity in IL-32γ-stimulated THP-1 cells was measured by FLISP, which is an experimental assay of intracellular chymotrypsin-like enzyme activity in living cells by using fluorescent-Labeled inhibitors of serine proteases. Photomicrography and fluorescence spectrometry revealed that addition of IL-32γ increased the fluorescent intensity in cultured THP-1 cells, indicating increased serine protease activity (Fig. 3a, b). Phenylmethanesulfonyl fluoride (PMSF), a general serine/cysteine protease inhibitor, significantly decreased the mRNA expressions and protein production of both TNFα and IFN-β induced by IL-32γ (Fig. 3c). When THP-1 cells were transfected with siRNA targeting PR3, levels of IL-32γ-induced TNFα and IFN-β declined markedly, while transfection with control siRNA did not show such effect, indicating that PR3 knockdown might mostly cancel IL-32-induced TNFα and IFN-β (Fig. 3d). PR3 thus appears indispensable for mRNA expression and protein production of TNFα and IFN-β in response to IL-32γ.

Activation of PR3 is indispensable for IL-32γ-induced TNFα and IFN-β mRNA expression and protein production.
Whether IL-32γ activated PR3 in cultured THP-1 cells was examined using FLISP, an experimental reagent to respond to serine protease activity. IL-32γ could activate serine proteases ((a), NC: negative control, FLISP: added only FLISP). Using fluorometry, IL-32γ was seen to stimulate serine protease activity as well (b). To examine the specific interaction of IL-32γ with PR3, levels of TNFα and IFN-β expressed in THP-1 cells were measured, when administered PMSF or transfected with siRNA targeting PR3. Both PMSF and siRNA of PR3 decreased IL-32γ-induced TNFα and IFN-β at both mRNA and protein levels (c, d). In particular, siRNA decreased levels of the two cytokines to basal levels while each RNAi negative control (ct-si) did not exhibit this effect. The ‘control’ signifies cells stimulated with IL-32γ and Lipofectamine 2000 (c, d). Levels of TNFα or IFN-β mRNA are shown as a proportion of levels from samples without IL-32γ stimulation, expressed as the mean ±SD for four independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
PAR2-TRIF and PAR2-Ras-Raf-1 pathways are chiefly responsible for transduction of IL-32γ signal to TNFα and IFN-β mRNA expression
As PAR2 has been shown to be activated by serine proteases such as PR3, interactions between IL-32γ and PAR2 or relevant downstream molecules were examined. IL-32γ-stimulated TNFα and IFN-β were completely abrogated when THP-1 cells were transfected with siRNA targeting PAR2, while transfection with control siRNA did not exert such effect (Fig. 4a). GW5074, an inhibitor of Raf-1, significantly diminished TNFα expression at both mRNA and protein levels by THP-1 cells, whereas mRNA expression or protein production of IFN-β was unchanged (Fig. 4b). Transfection of siRNA targeting TRIF resulted in reduced mRNA expression and protein production of both TNFα and IFN-β by THP-1 cells in response to IL-32γ, whereas no significant effect was observed with siRNA targeting TRIF-related adaptor molecule (TRAM) (Fig. 4c). These results indicate that IL-32γ induced TNFα using the two signaling arms of PAR2, including the Ras-Raf axis and TRIF-TRAF6 axis and induced IFN-β mRNA expression using only the TRIF axis.

PAR2-TRIF and PAR2-Ras-Raf-1 pathways are chiefly responsible for transduction of the IL-32γ signal to TNFα and IFN-β expression.
In THP-1 cells transfected with siRNA targeting PAR2, the mRNA and protein levels of IL-32γ-induced TNFα and IFN-β were decreased to basal levels (a). Transfection with RNAi negative control did not exhibit this effect. GW5074, a specific inhibitor of Ras-Raf signaling, suppressed IL-32γ-induced TNFα, but not IFN-β (b). IL-32γ was added to THP-1 cells transfected with siRNA targeting TRIF or TRAM. Both mRNA and protein levels of TNFα and IFN-β decreased with siRNA targeting TRIF, but not with siRNA targeting TRAM (c). The ‘control’ signifies cells stimulated with IL-32γ and Lipofectamine 2000 and ‘ct-si’ signifies RNAi negative control (a, c). Levels of TNFα and IFN-β mRNA are shown as a proportion of levels from samples without IL-32γ stimulation, with values expressed as the mean ±SD for four independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
Neutralization of TNFα markedly increases levels of IL-32γ-induced IFN-β
As TNFα is reportedly an inducer of type I IFN26, the interrelationship between TNFα and IFN-β (both of which can be induced by IL-32γ) was assessed using etanercept, a soluble TNFα receptor fusion protein used widely for treating inflammatory arthropathy and inflammatory bowel disease in humans. Etanercept prominently upregulated the level of IFN-β mRNA and production induced by IL-32γ (Fig. 5a, p < 0.01). This indicated that TNFα induced by IL-32γ rather suppressed IFN-β expression induced by IL-32γ. Meanwhile, since type I interferon has been reported to suppress the induction of TNFα27, Mouse Monoclonal Antibody against Human Interferon-Alpha/Beta Receptor 1 (IFNAR) and IL-32γ were simultaneously added to THP-1 cells in culture. This combination of IFNAR did not affect the level of TNFα mRNA expression and protein production induced by IL-32γ, whereas the level of TNFα mRNA and protein production constitutively expressed in THP-1 cells was increased by addition of IFNAR (Fig. 5b, c). Collectively, in THP-1 cells, IFN-β was capable of suppressing constitutive TNFα, but did not influence TNFα induced upon IL-32γ-stimulation.

Neutralization of TNFα markedly increases levels of IL-32γ-induced IFN-β.
When THP-1 cells were stimulated with IL-32γ simultaneously with etanercept, a specific soluble TNFα receptor, levels of IFN-β expression were prominently increased (a). IFNAR significantly increased levels of TNFα constitutively expressed in THP-1 cells (b). However, IFNAR had no effect on levels of IL-32γ-induced TNFα (c). Levels of TNFα or IFN-β mRNA are shown as a proportion of levels from samples without IL-32γ stimulation, with values expressed as the mean ±SD of four independent experiments (a,b). **p < 0.01, ***p < 0.001.
Protein kinase C (PKC) subtypes regulate expression of TNFα and IFN-β mRNA induced by IL-32γ
PKC is reportedly involved in TLR and PAR2 signaling pathways28,29. Several PKC isotypes have been identified, including conventional PKC (α,β and γ), novel PKC (δ, ε, et al.) and atypical PKC (ζ, et al.). Bisindolylmaleimide (BIS), a pan-PKC inhibitor and rottlerin, a specific inhibitor for PKCδ, significantly downregulated IL-32γ-induced TNFα and IFN-β (p < 0.05), whereas Gö6976, a PKCα/β inhibitor, did not (Fig. 6a). Likewise, siRNA against PKCδ decreased levels of IL-32γ-induced TNFα and IFN-β (p < 0.05), whereas siRNA targeting PKCε conversely increased levels of these cytokines (Fig. 6b, p < 0.05). TRAM adaptor with GOLD domain (TAG) is a splicing variant of TRAM and is known as a TRAM inhibitor30,31. IL-32γ stimulated mRNA expression of TAG and PMA markedly augmented TAG expression (Fig. 6c).

PKC subtypes regulate expression of TNFα and IFN-β mRNA induced by IL-32γ.
PKCs have been reported to mediate TLR and PAR2 signaling pathways. Bisindolylmaleimide (BIS) is a pan-PKC inhibitor and Gö6976 and rottlerin are specific inhibitors for conventional PKC and PKCδ, respectively. BIS and rottlerin significantly decreased levels of IL-32γ-induced TNFα and IFN-β, whereas Gö6976 displayed no significant effect on levels of these cytokines (a). Transfection of siRNA targeting PKCδ decreased levels of IL-32γ-induced TNFα and IFN-β, whereas siRNA targeting PKCε increased levels of TNFα and IFN-β, while each control did not exhibit this effect (b). IL-32γ alone induced TAG and PMA prominently accelerated IL-32γ-induced TAG mRNA expression (c). The ‘control’ signifies cells stimulated with IL-32γ and Lipofectamine 2000 and ‘ct-si’ signifies RNAi negative control (b). Levels of TNFα, IFN-β and TAG mRNA are shown as a proportion of each mRNA level expressed in the absence of IL-32γ stimulation, with values given as the mean ±SD for four independent experiments. *p < 0.05, **p < 0.01.
Discussion
Over the 48 h stimulation with LPS, mRNA expressions of both TNFα and IFN-β were upregulated in THP-1 cells with an apparently bimodal pattern, although the second peak of these expressions disappeared upon suppression of IL-32γ by RNA interference. Given that LPS was a potent inducer of IL-32γ among a variety of PAMPs, this second peak appears attributable to LPS-induced IL-32γ. Actually, downstream signaling of LPS via TLR4 has been reported to use two distinct arms, MyD88 and TRIF. Both signaling pathways commonly mediate activation of NF-κB and MAPKs, with MyD88 for early-phase activation32 and TRIF for late-phase activation33,34. According to most studies in the literature, the expression pattern of TNFα is bimodal due to MyD88 and TRIF signaling arms and TNFα expression ordinarily terminates within 6 h after LPS stimulation. We therefore believe that bimodal TNFα expression persisting over 48 h after LPS-stimulation is attributable to LPS-induced IL-32γ.
PR3 has been reported as a binding protein of IL-3221 and has been speculated to activate PAR2, which may play a key role in exhibiting IL-32 bioactivity35. Recent works have clarified in vivo roles of PAR2 as a sensor for both endogenous and exogenous serine proteases to maintain immune homeostasis36,37. However, no reports have dealt with the detailed interactions between IL-32, PR3 and PAR2. The present study demonstrated that IL-32γ clearly upregulated serine protease activity and expressions of both TNFα and IFN-β mRNA induced by IL-32γ were canceled by serine/cysteine protease inhibitor, PMSF or siRNA targeting PR3. These two cytokine expressions were also completely blocked by siRNA targeting PAR2. Increased PR3 activity and subsequent activation of PAR2 are thus necessary for IL-32γ to exhibit its biological activities (Fig. 7). Past evidence regarding PR3-PAR2 interactions23,24 and the receptor-ligand relationship of IL-32 and PR321 corroborate our results. Recently, one investigator has reported that IL-32α and β are bound to integrin-αVβ3 with their own RGD motif, so integrin represents a candidate for the receptor of intracellular IL-3238. In our experimental condition with THP-1 cells, however, cyclo-RGDfV, an inhibitor of integrin-αVβ3, did not affect the bioactivity of exogenous IL-32γ (data not shown; Fig. S5).

Schematic representation of the LPS-TLR4 and IL-32-PR3-PAR2 signaling pathways.
Once LPS from Gram-negative bacteria is recognized by TLR4, the TLR4-MyD88 pathway is triggered and early-phase TNFα expression is initiated through the activation of NF-κB and MAPKs. Activation of PKCε results in activation of TRAM-TRIF, which is responsible for late-phase TNFα and type I interferon (i.e., IFN-α/β) expression. Concurrently, IL-32γ binds to PR3 and activates PAR2. PAR2-Ras and -TRIF pathways potentially induce TNFα expression and the latter pathway in particular induces type I interferon expression via IRF3/7. At that time, TAG is synthesized to terminate TLR4-TRIF signaling, with TRIF preferentially recruited to PAR2 instead. Following LPS-induced TNFα expression mainly through TLR4, LPS-induced IL-32γ-PAR2 signaling gradually increases type I interferon expression, which potentially translates innate to adaptive immunity, or ceases a series of LPS-induced acute inflammation.
Previous studies have shown that the Ras-Raf signaling pathway and TRIF pathway are important for downstream signaling of PAR2, both of which ultimately lead to induction of TNFα (Fig. 7)39,40,41. In the present study, GW5074, the specific inhibitor for Raf-1 and siRNA targeting TRIF significantly diminished biological activities of IL-32γ, reflecting the existence of IL-32-PAR2 interactions in THP-1 cells. The putative IL-32γ-PAR2 axis is further supported by the fact that increased intracellular calcium levels following IL-32γ stimulation42 are compatible with the ability of PAR2 to cause intracellular calcium mobilization via G-protein39,40,41.
Without IL-32γ-induced TNFα production, IL-32γ might induce type I interferon more prominently, since our results revealed that etanercept markedly potentiated IFN-β mRNA expression induced by IL-32γ. On the other hand, addition of IFNAR did not affect TNFα production induced by IL-32γ, but instead increased levels of TNFα constitutively produced by THP-1 cells. In the context of bacterial or viral infection, the increment of TNFα expression is a very early event, followed by gradual increases in the level of type I interferon with decreasing levels of TNFα. Type I interferon has been shown to be essential for both Th1 cell polarization and antibody production (i.e., Th2), thus shaping adaptive immunity. Taken together with the present results that LPS is the strongest inducer of IL-32γ and that LPS-induced IL-32γ ultimately induces type I interferon production, IL-32γ appears to play a role in the transition from innate immunity to antigen-specific adaptive immunity in combination with IL-1, IL-6, IL-12, IL-18 and IL-23. TAG, a splice variant of TRAM, acts like a dominant-negative inhibitor of TRAM and has been reported to negatively regulate the TRAM-TRIF pathway31. IL-32γ-induced TAG presumably blocks the TRAM-TRIF axis and favors the PAR2-TRIF axis, corroborating the fact that IL-32γ-induced TNFα and IFN-β mRNA expressions were predominantly mediated by TRIF, but not by TRAM in our study. When TLR4 is stimulated with LPS, IL-32γ-induced TAG might contribute to termination of the LPS-TLR4-TRAM-TRIF axis and instead, augment the IL-32γ-PAR2-TRIF axis for the transition from innate to adaptive immunity.
The significance of the IL-32γ-PAR2-TRIF axis should be also recognized as an alternative signaling pathway to the LPS-TLR4-TRIF axis in shaping adaptive immunity. Potential situations with bacterial infection occasionally endanger the LPS-TLR4 system, in what has been perceived as an endotoxin tolerance. Endotoxin tolerance has been shown to suppress LPS-inducible TLR4-TRIF and TRIF-TRAF associated NF-κB activator (TANK)-binding kinase (TBK)1 associations followed by inactivation of IRF-343. Cario et al. reported that trypsin-like proteases derived from bacteria cause extensive proteolysis of myeloid differentiation factor 2 (MD-2) which contains multiple trypsin cleavage sites, leading to impaired LPS recognition of TLR444. Dysfunction of the TLR4 signaling pathway ultimately attenuates MyD88- and TRIF-dependent proinflammatory cytokine expression by macrophages, with unchanged or increased production of anti-inflammatory cytokines and increased incidence of secondary infections45. Impaired trafficking of TLR4 occasionally influences recruitment of TRIF. TLR4 mobilization from endosomes to phagosomes is a process necessary for the recruitment of TRIF and subsequent induction of type I interferon, whereas PAR2 recruits TRIF on the cell surface46. PAR2-TRIF engagement is advantageous to certain bacterial infection with disrupted endocytosis and escape from host defenses47. Furthermore, IL-32γ extracellularly modulates PAR2 by binding to PR3. The IL-32-PAR2-TRIF axis might thus be evolutionarily gained against the above-mentioned multiple types of endotoxin tolerances, including inactivated TBK1, trypsin cleavage of MD-2 and impaired TLR4 trafficking.
PAR2 couples to and activates G-proteins and leads to induction of canonical PLC/PKC signaling29,38. Among various PKC subtypes, PKCδ is reportedly involved in TLR signaling through its interaction with TIR domain containing adaptor protein (TIRAP), upstream of MyD8828,48. PKCε phosphorylates TRAM in the presence of MyD8849. Our study revealed that siRNA targeting PKCδ downregulated IL-32γ-induced TNFα and IFN-β; in contrast, siRNA targeting PKCε led to upregulation of these cytokines. Both TLR4 and PAR2 commonly use TRIF for each downstream signaling, suggesting the feasibility of competitive recruitment of TRIF between these two PRRs. Inhibition of PKCδ might result in activation of PKCε, increasing the proportion of TRIF associated with TLR4 but not with PAR2 and subsequently reducing the levels of IL-32γ-induced TNFα and IFN-β. In contrast, inhibition of PKCε increases the proportion of TRIF associated with PAR2, followed by increment of IL-32γ-induced TNFα and IFN-β. PKC subtypes including δ and ε therefore control the balance between the TLR4-TRIF and PAR2-TRIF axes and PKCδ in particular favors the PAR2-TRIF axis through increased expression of TAG (Fig. 7).
In summary, once LPS from Gram-negative bacteria is recognized by TLR4, TLR4-MyD88 pathway is triggered and early-phase TNFα expression is initiated through the activation of NF-κB and MAPKs (Fig. 7). As for differential activation of PKC isoforms, PKCδ is activated first, followed by PKCε, resulting in activation of the TRAM-TRIF pathway to contribute to late-phase TNFα expression. Concurrently, IL-32γ induced by TLR4 agonist such as LPS binds to PR3 and activates PAR2. The PAR2-Ras and -TRIF pathways induce TNFα expression and the latter pathway particularly induces type I interferon expression via IRF3/7. At that time, PKCδ is activated and PKCε is suppressed50 and TAG is synthesized to terminate TLR4-TRIF signaling, whereby TRIF is preferentially recruited to PAR2 instead. Following LPS-induced TNFα expression mainly through TLR4, LPS-induced IL-32γ-PAR2 signaling gradually increases type I interferon expression to translate innate to adaptive immunity, or to terminate the cascade of LPS-induced acute inflammation. Given that IL-32 exists only in higher mammals, the IL-32-PAR2-TRIF axis may have been gained during the evolution of mammalian immune systems in order to function not only as an extracellular sensor of bacterial and autologous proteases, but also as an interface between innate and adaptive immunity.
Methods
Reagents
Recombinant human IL-32γ protein (rIL-32γ) was purchased from R&D Systems (Minneapolis, MN, USA). Human rIL-32γ was tested for endotoxin contamination with Limulus ESII test, which was purchased from Wako Pure Chemical Industries (Osaka, Japan). LPS from Escherichia coli 0111:B4, zymosan A from Saccharomyces cerevisiae, poly (I:C), PMSF, PMA, Gö6976 and rottlerin were purchased from Sigma-Aldrich (St. Louis, MO, USA). Imiquimod, TLR7 agonist, was purchased from Merck Millipore (Billerica, MA, USA). CpG-B DNA (human/mouse), a TLR9 agonist, was purchased from Hycult Biotech (Uden, the Netherlands). GW5074 was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Etanercept was obtained from Wyeth Japan (Tokyo, Japan). IFNAR was purchased from Pestka Biomedical Laboratories (Piscataway, NJ, USA). BIS was purchased from Cell Signaling Technology Japan (Tokyo, Japan). PMSF, BIS, Gö6976, rottlerin and GW5074 were dissolved in 100% dimethyl sulfoxide (DMSO) at 100 mM and stored in aliquots at −30°C. Before use in cell culture, these agents were diluted with the medium to a final DMSO concentration of ≤0.05%.
Real-time polymerase chain reaction (PCR) analysis
Total RNAs were isolated from each sample by RNAiso plus® (Takara Bio, Kyoto, Japan) and cDNAs were synthesized from total RNAs using a PrimeScript RT® reagent kit (Takara Bio). Real-time PCR was performed using SYBR Premix ExTaq II® (Takara Bio) with a DICE thermal cycler® (Takara Bio), according to the instructions from the manufacturer. Results were normalized to glyceraldhyde-3-phosphate dehydrogenase (GAPDH) as the fold change compared with samples. The primer sequences used in this study are presented in Table 1.
ELISA
IL-32-specific enzyme-linked immunosorbent assay (ELISA) (Human Interleukin 32 ELISA Kit) was purchased from Cusabio (Wuhan, China) and TNFα-specific ELISA (Quantikine Human TNFα) was purchased from R&D Systems (Minneapolis, MN, USA). IFN-β-specific ELISA (VeriKine-HS Human Interferon Beta Serum ELISA Kit) was purchased from PBL Interferon Source (Piscataway, NJ, USA).
Cell culture
THP-1 cells were obtained from American Type Culture Collection (Manassas, VA, USA) and cultured in RPMI1640 medium (Life Technologies, CA, USA) containing 10% fetal calf serum (FCS), 100 U/ml penicillin and 100 μg/ml streptomycin.
Differentiation of THP-1 cells
Differentiation of THP-1 cells to macrophages was triggered by addition of PMA (50 ng/ml), followed by overnight incubation. After washing twice, the cells were incubated with RPMI1640 for 48 h. These differentiated THP-1 cells were subjected to the all experiments.
FLISP
FLISP® was reagent for measurement of the activities of serine proteases and purchased from Immunochemistry Technologies (Bloomington, MN, USA). Experiments with THP-1 were performed according to the instructions from the manufacturer and fluorescence spectrometry was undertaken using Power Scan HT (DS Pharma Biomedical, Osaka, Japan).
Construction and transfection of small interfering RNA (siRNA)
Stealth RNA interference (RNAi) against multiple molecules and each Stealth RNAi-negative control, which was one or a few bases different from original RNAi sequences, were purchased from Life Technologies Japan (Tokyo, Japan). Sequences of the Stealth RNAi used in this study are presented in Supplementary Table 1. THP-1 cells were transfected with 40 nM siRNA using Lipofectamine 2000 (Life Technologies) according to the instructions from the manufacturer and then treated at 24 h post-transfection. Specific gene silencing was confirmed by real-time PCR (Figs. S1–S4).
Stimulation of cells
THP-1 cells (1 × 105 cells) were cultured with LPS (100 ng/ml or 1 μg/ml), zymosan (10 or 100 μg/ml), poly (I:C) (10 or 100 μg/ml), imiquimod (10 or 100 μg/ml) and CpG-B DNA (10 or 100 μM) for 6 h, then the amount of IL-32 was measured by real-time PCR. As to cells culturing with LPS, zymosan and poly (I:C), the amount of IL-32 protein was measured by specific ELISA. THP-1 cells (1 × 105 cells) were cultured with LPS (100 ng/ml) for 6, 12, or 24 h and the amount of produced IL-32 was measured by real-time PCR and specifc ELISA.
THP-1 cells (1 × 105 cells) were incubated with rIL-32γ or LPS for 3, 6, 12, 24, or 48 h and the amount of TNFα or IFN-β was measured by real-time PCR and specific ELISA. THP-1 cells (1 × 105 cells) transfected with siRNA against IL-32 were incubated with LPS for 3, 6, 12, 24, or 48 h and the amount of TNFα or IFN-β was measured by real-time PCR and ELISA. THP-1 cells (1 × 105 cells) were cultured with rIL-32γ and/or PMSF (1 mM) or GW5074 (5 μM) for 6 h and the amount of TNFα or IFN-β was measured by real-time PCR and ELISA. THP-1 cells (1 × 105 cells) transfected with siRNA against PR3 or PAR2 or control siRNA were incubated with rIL-32γ for 6 h and the mRNA and protein levels of TNFα or IFN-β were measured by real-time PCR and ELISA, respectively.
THP-1 cells (1 × 105 cells) were cultured with rIL-32γ and/or etanercept (100 μg/ml) for 6 h and the amount of IFN-β was measured by real-time PCR and ELISA. THP-1 cells (1 × 105 cells) were cultured with or without rIL-32γ and/or IFNAR (2 μg/ml) for 6 h and the amount of TNFα was measured by real-time PCR and ELISA. THP-1 cells (1 × 105 cells) were incubated with PMA (100 ng/ml), BIS (10 μM), Gö6976 (1 μM), or rottlerin (10 μM) 30 min before rIL-32γ stimulation, then the amount of TNFα or IFN-β was measured by real-time PCR and ELISA after 6 h incubation. THP-1 cells (1 × 105 cells) were cultured with rIL-32γ and/or PMA (100 ng/ml) and the level of TAG was measured by real-time PCR.
Statistical analysis
Results are reported as mean ± standard deviation (SD). Statistical analysis was undertaken using a two-tailed Student's t-test or one-way ANOVA. Differences were considered statistically significant at the p < 0.05 level. All experiments were performed in four times.
References
Dahl, C. A., Schall, R. P., He, H. L. & Cairns, J. S. Identification of a novel gene expressed in activated natural killer cells and T cells. J Immunol 15, 597–603 (1992).
Kim, S. H., Han, S. Y., Azam, T., Yoon, D. Y. & Dinarello, C. A. Interleukin-32: a cytokine and inducer of TNF-alpha. Immunity 22, 131–142 (2005).
Choi, J. D. et al. Identification of the most active interleukin-32 isoform. Immunology 126, 535–542 (2009).
Goda, C. et al. Involvement of IL-32 in activation-induced cell death in T cells. Int Immunol 18, 233–240 (2006).
Netea, M. G. et al. Mycobacterium tuberculosis induces interleukin-32 production through a caspase-1/IL-18/interferon-gamma-dependent mechanism. PLoS Med 3, e277 (2006).
Bai, X. et al. IL-32 is a host protective cytokine against Mycobacterium tuberculosis in differentiated THP-1 human macrophages. J Immunol 184, 3830–3840 (2010).
Rasool, S. T. et al. Increased level of IL-32 during human immunodeficiency virus infection suppresses HIV replication. Immunol Lett 117, 161–167 (2008).
Li, W. et al. IL-32: a host proinflammatory factor against influenza viral replication is upregulated by aberrant epigenetic modifications during influenza A virus infection. J Immunol 185, 5056–5065 (2010).
Smith, A. J. et al. The immunosuppressive role of IL-32 in lymphatic tissue during HIV-1 infection. J Immunol 186, 6576–6584 (2011).
Shioya, M. et al. Epithelial overexpression of interleukin-32alpha in inflammatory bowel disease. Clin Exp Immunol 149, 480–486 (2007).
Calabrese, F. et al. IL-32, a novel proinflammatory cytokine in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 178, 894–901 (2008).
Nold, M. F. et al. Endogenous IL-32 controls cytokine and HIV-1 production. J Immunol 181, 557–565 (2008).
Nishida, A. et al. Phosphatidylinositol 3-kinase/Akt signaling mediates interleukin-32alpha induction in human pancreatic periacinar myofibroblasts. Am J Physiol Gastrointest Liver Physiol 294, G831–G838 (2008).
Mun, S. H. et al. Tumor necrosis factor alpha-induced interleukin-32 is positively regulated via the Syk/protein kinase Cdelta/JNK pathway in rheumatoid synovial fibroblasts. Arthritis Rheum 60, 678–685 (2009).
Ko, N. Y. et al. Interleukin-32α production is regulated by MyD88-dependent and independent pathways in IL-1β-stimulated human alveolar epithelial cells. Immunobiology 216, 32–40 (2011).
Alsaleh, G. et al. Innate immunity triggers IL-32 expression by fibroblast-like synoviocytes in rheumatoid arthritis. Arthritis Res Ther 12, R135 (2010).
Pan, X. et al. Interleukin-32 expression induced by hepatitis B virus protein X is mediated through activation of NF-κB. Mol Immunol 48, 1573–1577 (2011).
Nakayama, M. et al. Enhanced susceptibility to lipopolysaccharide-induced arthritis and endotoxin shock in interleukin-32 alpha transgenic mice through induction of tumor necrosis factor alpha. Arthritis Res Ther 14, R120 (2012).
Netea, M. G. et al. IL-32 synergizes with nucleotide oligomerization domain (NOD) 1 and NOD2 ligands for IL-1beta and IL-6 production through a caspase 1-dependent mechanism. Proc Natl Acad Sci U S A 102, 16309–16314 (2005).
Heinhuis, B. et al. IL-32 gamma and Streptococcus pyogenes cell wall fragments synergise for IL-1-dependent destructive arthritis via upregulation of TLR-2 and NOD2. Ann Rheum Dis 69, 1866–1872 (2010).
Novick, D. et al. Proteinase 3 is an IL-32 binding protein. Proc Natl Acad Sci U S A 103, 3316–3321 (2006).
Uehara, A., Sugawara, S., Muramoto, K. & Takada, H. Activation of human oral epithelial cells by neutrophil proteinase 3 through protease-activated receptor-2. J Immunol 169, 4594-4603 (2002).
Csernok, E., Holle, J. U. & Gross, W. L. Proteinase 3, protease-activated receptor-2 and interleukin-32: linking innate and autoimmunity in Wegener's granulomatosis. Clin Exp Rheumatol 26, S112–S117 (2008).
Jiang, B. et al. The role of proteinase 3 (PR3) and the protease-activated receptor-2 (PAR-2) pathway in dendritic cell (DC) maturation of human-DC-like monocytes and murine DC. Clin Exp Rheumatol 28, 56–61 (2010).
Witko-Sarsat, V. et al. A large subset of neutrophils expressing membrane proteinase 3 is a risk factor for vasculitis and rheumatoid arthritis. J Am Soc Nephrol 10, 1224–1233 (1999).
Yamamoto, M. et al. TRAM is specifically involved in the Toll-like receptor 4-mediated MyD88-independent signaling pathway. Nat Immunol 4, 1144–1150 (2003).
Palsson-McDermott, E. M. et al. TAG, a splice variant of the adaptor TRAM, negatively regulates the adaptor MyD88-independent TLR4 pathway. Nat Immunol 10, 579–586 (2009).
Kawai, T. & Akira, S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 11, 373–384 (2010).
Kawai, T., Adachi, O., Ogawa, T., Takeda, K. & Akira, S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity 11, 115–122 (1999).
Kaisho, T., Takeuchi, O., Kawai, T., Hoshino, K. & Akira, S. Endotoxin-induced maturation of MyD88-deficient dendritic cells. J Immunol 166, 5688–5694 (2001).
Dinarello, C. A. & Kim, S. H. IL-32, a novel cytokine with a possible role in disease. Ann Rheum Dis 65, iii61–iii64 (2006).
Park, M. K. et al. Protease-activated receptor 2 is involved in Th2 responses against Trichinella spiralis infection. Korean J Parasitol 49, 235–243 (2011).
Chung, W. O. et al. Interplay of protease-activated receptors and NOD pattern recognition receptors in epithelial innate immune responses to bacteria. Immunol Lett 131, 113–119 (2010).
Heinhuis, B. et al. Interleukin 32 (IL-32) contains a typical α-helix bundle structure that resembles focal adhesion targeting region of focal adhesion kinase-1. J Biol Chem 287, 5733–5343 (2012).
Macfarlane, S. R., Seatter, M. J., Kanke, T., Hunter, G. D. & Plevin, R. Proteinase-activated receptors. Pharmacol Rev 53, 245–282 (2001).
Goon Goh, F. et al. G-protein-dependent and -independent pathways regulate proteinase-activated receptor-2 mediated p65 NFkappaB serine 536 phosphorylation in human keratinocytes. Cell Signal 20, 1267–1274 (2008).
Rothmeier, A. S. & Ruf, W. Protease-activated receptor 2 signaling in inflammation. Semin Immunopathol 34, 133–149 (2012).
Jeong, H. J., Han, N. R., Moon, P. D., Kim, M. H. & Kim, H. M. Intracellular calcium level is upregulated by interleukin-32 in auditory cells. Cytokine 53, 153–157 (2011).
Yarilina, A. & Ivashkiv, L. B. Type I interferon: a new player in TNF signaling. Curr Dir Autoimmun 11, 94–104 (2010).
Benveniste, E. N. & Qin, H. Type I interferons as anti-inflammatory mediators. Sci STKE 416, pe70 (2007).
Piao, W. et al. Endotoxin tolerance dysregulates MyD88- and Toll/IL-1R domain-containing adapter inducing IFN-beta-dependent pathways and increases expression of negative regulators of TLR signaling. J Leukoc Biol. 86, 863–875 (2009).
Cario, E. et al. Trypsin-sensitive modulation of intestinal epithelial MD-2 as mechanism of lipopolysaccharide tolerance. J Immunol 176, 4258–4266 (2006).
Cavaillon, J. M. & Adib-Conquy, M. Bench-to-bedside review: endotoxin tolerance as a model of leukocyte reprogramming in sepsis. Crit Care 10, 233 (2006).
Rallabhandi, P. et al. Analysis of proteinase-activated receptor 2 and TLR4 signal transduction: a novel paradigm for receptor cooperativity. J Biol Chem 283, 24314–24325 (2008).
Taxman, D. J. et al. Porphyromonas gingivalis mediates inflammasome repression in polymicrobial cultures through a novel mechanism involving reduced endocytosis. J Biol Chem 287, 32791–32799 (2012).
van der Merwe, J. Q., Moreau, F. & MacNaughton, W. K. Protease-activated receptor-2 stimulates intestinal epithelial chloride transport through activation of PLC and selective PKC isoforms. Am J Physiol Gastrointest Liver Physiol 296, G1258–1266 (2009).
Loegering, D. J. & Lennartz, M. R. Protein kinase C and toll-like receptor signaling. Enzyme Res 2011, 537821; 10.4061/2011/537821 (2011).
Wermuth, P. J., Addya, S. & Jimenez, S. A. Effect of protein kinase C delta (PKC-δ) inhibition on the transcriptome of normal and systemic sclerosis human dermal fibroblasts in vitro. PLoS One 6, e27110 (2011).
McGettrick, A. F. et al. Trif-related adapter molecule is phosphorylated by PKCε during Toll-like receptor 4 signaling. Proc Natl Acad Sci U S A 103, 9196–9201 (2006).
Pears, C. J. et al. Differential roles of the PKC novel isoforms, PKCδ and PKCε, in mouse and human platelets. PLoS One 3, e3793 (2008).
Acknowledgements
This work was partially supported by a Grant-in-aid for Scientific Research (C) (#21591959) from the Japan Society for the Promotion of Science.
Author information
Authors and Affiliations
Contributions
M.N. carried out molecular experiments, performed the statistical analyses and drafted the manuscript. Y.N. conceived and designed the study and edited the manuscript. T.K. taught the experimental procedure and advised M.N. on this study. Y.T. was involved in the conception and design of the study. H.I., Y.T. and T.M. supervised the study design and provided valuable advice to M.N. All authors approved the final version of the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary manuscript
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Nakayama, M., Niki, Y., Kawasaki, T. et al. IL-32-PAR2 axis is an innate immunity sensor providing alternative signaling for LPS-TRIF axis. Sci Rep 3, 2960 (2013). https://doi.org/10.1038/srep02960
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep02960
This article is cited by
-
High serum proteinase-3 levels predict poor progression-free survival and lower efficacy of bevacizumab in metastatic colorectal cancer
BMC Cancer (2024)
-
Assessment of interleukin 32 as a novel biomarker for non-alcoholic fatty liver disease
Egyptian Liver Journal (2022)
-
MicroRNA-29 family expression and its relation to antiviral immune response and viro-immunological markers in HIV-1-infected patients
BMC Infectious Diseases (2015)
-
Proteinase 3 Induces Neuronal Cell Death Through Microglial Activation
Neurochemical Research (2015)
-
Interleukin-32 isoforms: expression, interaction with interferon-regulated genes and clinical significance in chronically HIV-1-infected patients
Medical Microbiology and Immunology (2014)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
