
Abstract
Quorum sensing (QS) is a population-dependent mechanism for bacteria to synchronize social behaviors such as secretion of virulence factors. The enzymatic interruption of QS, termed quorum quenching (QQ), has been suggested as a promising alternative anti-virulence approach. In order to efficiently identify QQ bacteria, we developed a simple, sensitive and high-throughput method based on the biosensor Agrobacterium tumefaciens A136. This method effectively eliminates false positives caused by inhibition of growth of biosensor A136 and alkaline hydrolysis of N-acylhomoserine lactones (AHLs), through normalization of β-galactosidase activities and addition of PIPES buffer, respectively. Our novel approach was successfully applied in identifying QQ bacteria among 366 strains and 25 QQ strains belonging to 14 species were obtained. Further experiments revealed that the QQ strains differed widely in terms of the type of QQ enzyme, substrate specificity and heat resistance. The QQ bacteria identified could possibly be used to control disease in aquaculture.
Similar content being viewed by others

Introduction
Quorum sensing (QS) is a population-dependent mechanism for bacteria to communicate, regulate gene expression and synchronize social behaviors such as biofilm formation1, bioluminescence2,3 and secretion of virulence factors4. N-acylhomoserine lactones (AHLs) are used by Gram-negative Proteobacteria as autoinducers for intraspecies communications and are composed of a homoserine lactone (HSL) ring carrying acyl chains of 4 to 18 carbons with or without a modification at the C3 position5,6,7,8. AHL dependent QS was first discovered in two luminescent marine bacteria, Vibrio fischeri and V. harveyi, several decades ago2,3 and has been recognized to play an important role in the expression of virulence factors in many pathogens including Vibrio6,7,9. For example, the LuxM/N system of V. anguillarum was shown to use 3-hydroxy-C6-HSL and C6-HSL as signals to regulate the expression of virulence factors such as metalloprotease7.
Interfering with the QS system was suggested as a promising alternative to antibiotics because the expression of virulence factors could be effectively turned off without imposing a ‘life-or-death’ selective pressure on the pathogens10. This strategy would also be less likely to induce resistance. Interference can occur at any step of the QS circuit: production of signal, release of signal molecules, recognition of signal molecules by receptors and activation of target genes11. Interference can be due to small molecule antagonists12 or signal degrading enzymes13,14,15. In the latter case the term “quorum quenching” (QQ) is used. One group of small molecule antagonists, i.e., halogenated furanones, which were first found in Delisea pulchra, could effectively inactivate QS systems and reduce the virulence of many bacterial pathogens16. QQ enzymes were discovered in a wide range of bacteria and were classified into three major types according to their enzymatic mechanisms: AHL lactonase (lactone hydrolysis)13, AHL acylase (amidohydrolysis)14 and AHL oxidase and reductase (oxidoreduction)15. It has been demonstrated that the expression of QQ enzymes in plant pathogens could reduce their virulence13 and that AHL-degrading microbial enrichment cultures could improve the larval survival of brine shrimp17 and turbot18 in routine culture and protect giant freshwater prawn larvae from V. harveyi infection19. The identification of QQ bacteria from indigenous microbial communities of various environments could hence provide us with new agents for anti-virulence strategies to control disease caused by QS dependent pathogens and/or QS dependent host-microbial interactions.
However it is a challenge to efficiently identify QQ bacteria from hundreds or thousands of bacterial isolates. At present, biosensors are widely employed for identifying QQ bacteria because of high sensitivity and a wide-range detectability. The AHL biosensors express reporter genes (such as lacZ, gfp or luxCDABEG) in response to specific AHLs20. The Agrobacterium tumefaciens derived strains21,22 can detect a broad range of AHLs by expressing β-galactosidase (encoded by lacZ), which can be quantitatively measured by using various substances, i.e., colorimetric substances (X-gal and ONPG), chemiluminescent substances and fluorescent substances23. Although the ONPG assay24 is a standard method to monitor β-galactosidase, there are two major inherent drawbacks in this assay. Firstly, the sensitivity of ONPG detection (2-nitrophenyl β-D-galactopyranoside) is lower than that of X-gal (5-bromo-4-chloro-3-indolyl β-D-galactopyranoside)23. Secondly, since ONPG cannot cross intact membranes24, the biosensor cells need to be permeabilized, which is time and labor-consuming when manipulating many samples simultaneously. The X-gal assay can overcome these two drawbacks and the final blue products resulting from β-galactosidase activity, 5,5′-dibromo-4,4′-dichloro-indigo (indigo), can be quantified by measuring its absorbance at 630 nm25. However, it is a challenge to quantify the amount of indigo in a whole-cell assay, because the turbidity of the A. tumefaciens cells also contribute to the OD630 read-out. Although the quantification of β-galactosidase activity in whole-cell liquid X-gal assay could be improved by dissolution of indigo with DMSO26, this is also time and labor-consuming for high-throughput identifying QQ bacteria.
In the present study, we developed a simple, sensitive and high-throughput method for identifying QQ bacteria. In this assay, β-galactosidase activities can be accurately quantified by absorbance measurements followed by normalization to cell density of the biosensor. This allowed simultaneous measurement of large numbers of samples with a minimum of hands-on time, requiring merely standard equipment available in any research laboratory.
Results
The correction factor values a and b for the normalization of β-galactosidase activity
According to the Beer-Lambert law27, the measured absorbance can be expressed as the following formula: A = εbc (where A is the absorbance; ε is the extinction coefficient; b is the thickness of absorber; c is the concentration of absorber). Turbidity measurement can be also based on a derivation of Beer-Lambert law28. For a given absorber, the extinction coefficient is a function depending on wavelength of the light used and it is a constant at a specified wavelength. Therefore, for a given concentration of absorber (indigo and biosensor cells in our assay), the ratio of absorbances at different wavelengths is a constant and is equal to ελ1/ελ2 (correction factors a and b in our assay) in theory. As a result, the values of the correction factor a and b could be obtained through regression analysis of the absorbances of indigo and cells of biosensor A13621 with different concentrations at two different wavelengths. The choice of wavelengths was according to the absorbance spectra of indigo (Supplementary Fig. S1 online). Indigo showed relatively lower and higher absorbances at the wavelengths 492 nm and 630 nm respectively. Therefore, for the normalization of β-galactosidase activity, the wavelengths 492 nm and 630 nm were chosen to monitor β-galactosidase activities. In order to obtain the correction factor values a and b, OD492 and OD630 of a dilution series of biosensor A136 or indigo were measured. When plotting OD630 against OD492 of biosensor A136 (Fig. 1a) or OD492 against OD630 of indigo (Fig. 1b), a linear relationship was obtained (R2 > 0.99). The values of the correction factors a and b, 0.653 and 0.267, were obtained from Figure 1a and Figure 1b, respectively. Consequently, the full formula is:  .
.

Correction factors a and b.
OD630 and OD492 were measured with a series cell density of biosensor A136 (a) and 5,5′-dibromo-4,4′-dichloro-indigo (b). The linear correlation coefficients (R2) were shown in scatter diagrams and the slopes of the straight lines are the correction factor values a (a) and b (b), respectively. F-Tests were used to fit the best model.
Optimization of the A136 liquid X-gal assay
To obtain an appropriate protocol leading to reproducible results and a distinction between different concentrations of C6-HSL, the A136 liquid X-gal assay was optimized. To exclude false positives caused by alkaline degradation, different buffers were used to maintain a slightly acid pH of the reaction mixture. The pHs of bacterial cultures were ranged from 7.3 to 8.5 before co-incubation with AHLs. After addition of the PIPES and the MOPS buffers, the pHs of bacterial cultures both changed to slightly acid and increased insignificantly during the following co-incubation. No obvious alkaline degradation of C6-HSL was observed, indicating that both of the PIPES and the MOPS buffers were effective to prevent false positives. However, considering that the pKa of PIPES (6.71) is lower than the MOPS (7.01), the former was chosen for further experiments.
When an overnight culture of biosensor A136 (OD600 ≈ 2) was inoculated in the A136 X-gal assay solution with an inoculum size of 1%, the normalized β-galactosidase activities were proportional to concentrations of C6-HSL up to 2 μM and saturated quickly between 2 and 10 μM (Fig. 2). The normalized β-galactosidase activities were greatly affected by the incubation time and the inoculum size of the A136 X-gal assay solution. For a given concentration of C6-HSL, the lower the inoculum size was, the longer the incubation time needed (Supplementary Fig. S2 online).

Optimization of the C6-HSL concentration.
The normalized β-galactosidase activities increased linearly at lower AHL concentrations and gradually reached a saturation level at higher AHLs concentrations. Data were shown as mean ± SD.
The final optimized protocol for identification of QQ bacteria
A scheme of the final optimized protocol is shown in Figure 3. The normalized β-galactosidase activities were proportional to the concentrations of C6-HSL (0.1–0.5 μM) with good reproducibility when an overnight broth culture of A. tumefaciens A136 (OD600 ≈ 2) was inoculated in the A136 X-gal assay solution with an inoculum size of 1% and then incubated for 12 h. The normalization of β-galactosidase activity can effectively remove false positives caused by inhibition of growth of biosensor A136. Additionally, AHLs are unstable and easily inactived by lactonolysis in basic solution. Therefore, the PIPES stock solution (1 M, pH 6.7) was added in bacterial culture with a final concentration of 100 mM to maintain a slightly acid pH during the reaction, which effectively eliminated false positives caused by alkaline degradation of AHLs.

The final optimized protocol.
The reaction step was carried out in 8 PCR tubes for easy subsequent centrifugation. 96-well plates were used in the detection step to facilitatehigh-throughput measurements.
Comparison of the A136 liquid X-gal assay and the ONPG assay
The ONPG assay for measurement of β-galactosidase activities24 is a widely used method because of its accuracy, incentivating a comparison of the A136 liquid X-gal assay with the ONPG assay. The values of β-galactosidase activities measured by the A136 liquid X-gal assay were plotted against those obtained with the ONPG assay (Fig. 4a). The Pearson correlation coefficient was greater than 0.99, which indicated that the A136 liquid X-gal assay was as accurate as the ONPG assay. Furthermore, the normalized β-galactosidase activities had a linear relationship with the concentrations of C6-HSL in the range of 0.1 to 0.7 μM (Fig. 4b), which implied that the concentrations of C6-HSL can be quantified by the optimized A136 liquid X-gal assay. This also indicated that the rate or level of lacZ expression in biosensor A136 was quantitatively controlled by the concentrations of C6-HSL ranging from 0.1 to 0.7 μM.

Comparison of the A136 liquid X-gal assay with the ONPG assay.
a, The relationship between the A136 liquid X-gal assay and the ONPG assay. P value (two-tailed) < 0.0001 (The P value was computed by Pearson correlation calculations, n = 3 and alpha = 0.05). b, The relationships between the β-galactosidase activities and the concentration of C6-HSL. The R2 of linear regression for the A136 liquid X-gal assay ( ) and the ONPG assay (
) and the ONPG assay ( ) are 0.996 and 0.993, respectively. All the data were shown as mean ± SD.
) are 0.996 and 0.993, respectively. All the data were shown as mean ± SD.
Application of the A136 liquid X-gal assay for high-throughput identification of QQ bacteria
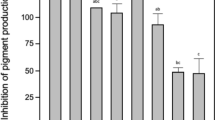
A total of 366 bacterial strains isolated from the gills, skin mucus and intestines of healthy flounder were screened for C6-HSL degrading activities by the A136 liquid X-gal assay. Subsequently, 25 strains that caused more than 40% reduction in the normalized β-galactosidase activities were selected for further identification and tests (Fig. 5).

The normalized β-galactosidase activities of the 25 QQ bacterial strains.
Each strain was compared with negative control using un-paired t-test (n = 3; two-tailed P value; ***P < 0.0001, **P < 0.001, *P = 0.0143).
Most of the 25 QQ strains possessed strong C6-HSL degrading activities except strains T171, T194, T202, Th15 and Th125, which showed relatively weaker activities (Fig. 5) and these results were confirmed by the CV026 plate assay29 (Table 1). Additionally, the pHs of reaction mixtures maintained below 7 during the whole process, which could ensure the veritable C6-HSL degrading activities but not alkaline degradation.
Based on 16S rRNA gene similarity these 25 QQ strains were classified into 14 species (Table 1) belonging to the classes Flavobacteriia, Gammaproteobacteria and Alphaproteobacteria. The relationships among the 25 strains and other representative QQ bacterial strains reported previously30 are shown in Figure 6 and Supplementary Figure S3 online. Twelve out of the 14 bacterial species identified in the present study have not been reported to bear AHL-degrading activities yet. Among the QQ strains, strains Th26, Th78, Th87, Th93 and Th106 (96.7%–96.8% similarity to Flaviramulus basaltis H35T) and T202 (96.6% similarity to Thalassomonas agariperforans M-M1T) could be novel species. Additionally, the majority of the 25 QQ strains were isolated from gills and intestines and only 2 strains were isolated from skin mucus (Table 1).

Neighbour-joining tree based on the 16S rRNA gene.
The relationships among the QQ bacterial strains from this study and the reported QQ bacteria were shown and the number after each strain is the accession number in NCBI database. Bootstrap coefficients below 50% were not shown. Scale bar, 0.05 substitutions per nucleotide position. *The 16S rRNA gene sequences of Pseudoalteromonas byunsanensis 1A0126157, Sphingopyxis witflariensis wtt2531 and Mycobacterium tuberculosis58 were not available, so P. byunsanensis FR119, S. witflariensis W-50 and M. tuberculosis H37Rv were used in the tree instead. **The detail of Bacillaceae branch was shown in Supplementary Figure S3 online.
Most of the bacterial strains were typical marine bacteria, except strains T51, T52, Th8 and Th21 (98.6–99.6% similarity to Sphingopyxis spp.) and strain Th20 (97.78% similarity to Novosphingobium tardaugens). QQ activity was previously identified in the species Sphingopyxis witflariensis isolated from tobacco rhizosphere31.
Further characterization of AHL-degrading activities of QQ bacteria
The heat resistance, type of QQ enzyme and substrate specificity of the 25 QQ bacteria were tested. The C6-HSL degrading activities in 10 out of 14 species were thermolabile (Table 1).
The lactone rings of AHLs could be opened by enzymolysis of AHL lactonase resulting with a hydroxyl group and a carboxyl group. The recyclization of opened lactone rings could be performed by acidification that the pH first approached the pK of the carboxyl group (pH 2) and then rings were closed with the hydroxyl group32. Therefore, the lactonase activities could be determined by acidification. Compared with lactonase AiiA and acylase containing strain P. aeruginosa ZJ551, the recovery of C6-HSL by acidification was observed in three species, including Flaviramulus spp., Olleya marilimosa and Tenacibaculum soleae (Supplementary Fig. S4), indicating these bacteria contain lactonase activities. Likewise, the recovery of C12-HSL was found in the three species when C12-HSL was co-incubated with bacterial cultures for 2 hours before acidification. However, when the co-incubation time was prolonged to 24 hours, the recovery of C12-HSL was only observed in O. marilimosa T168.
Most of the QQ strains degraded all the AHL substrates tested, except for strains T51, T52, T171, T194, Th15, Th30 and T202 that showed no activity against 3-oxo-C6-HSL (Table 1). The QQ activities of strains Th15 (98.20% similarity to Rhodobacter ovatus JA234T) and Th30 (97.4% similarity to Marivita byusanensis SMK-114T) showed strong activities against AHLs without oxo-group and strains T171 (99.4% similarity to Colwellia aestuarii SMK-10T), T194 (97.9% similarity to Salinimonas lutimaris) T202 and Th125 (99.6% similarity to Pseudoalteromonas prydzensis), showed strong activities against long-chain AHLs. These results indicated that a high diversity of AHL-degrading activities existing in these QQ strains.
Discussion
At present, the methods for identification of QQ bacteria could be classified to three types: minimum medium assay, plate inhibition assay and residual AHL measurement. The minimum medium assay is based on the capability of bacteria to utilize the AHLs as the sole source of carbon, nitrogen and energy. Despite the successful application in several studies33,34, the diversity and complexity of the identified QQ bacteria and enzymes were relatively low because the majority of the reported QQ bacteria cannot grow in the minimal medium but possess strong AHL-degrading activities30. The plate inhibition assay35 is based on agar diffusion of the QS-interfering molecules which would produce a visible inhibition zone in the plates when the co-incubated biosensor produced pigment or other phenotypes. This method is time and labor-consuming when testing hundreds of candidates and a strong QQ activity is required to generate detectable inhibition zones. The last method is generally carried out by the measurements of residual AHLs (direct measurement or measured by various biosensors) after given amount of AHLs reacted with candidate bacteria. Although the amount of residual AHLs can be quantified directly by colorimetry, GC-MS, LC and HPLC36, the insensitivity of colorimetry and the requirement for particular instruments of GC-MS, LC and HPLC limit the application in high-throughput identification of QQ bacteria. Therefore, various AHL biosensors were employed for identifying QQ bacteria and the A. tumefaciens derived strains21,22 can be used for detecting a broad range of AHLs. Nevertheless, the current method for detecting QQ bacteria with A. tumefaciens biosensors is still too cumbersome to allow high-throughput studies22 or too imprecise to be quantitative37,38.
The A136 liquid X-gal assay we developed in the present study is based on the principle of bio-measurement of the residual AHLs after reaction. Generally, there are two major improvements in the A136 liquid X-gal assay.
One of the improvements is the rapid, quantitative and high-throughput measurement of β-galactosidase activities. Although the ONPG assay is accurate to monitor β-galactosidase activities, the tedious processes result in poor efficiency and low throughput. On the contrary, in the A136 liquid X-gal assay, the quantitative measurement of β-galactosidase activity was achieved by normalization of indigo to the cell density of the biosensor which greatly simplified the procedure. Meanwhile, the 96-well plate layout enabled a one-step detection of β-galactosidase activity and therefore allowed a high-throughput identification of QQ bacteria from many candidates with a minimum of hands-on time. Moreover, this method could effectively eliminate false positives which might be due to inhibition of growth of biosensor A136 by metabolites of candidate QQ strains. Additionally, this improvement also allowed a continuous measurement of β-galactosidase activities while the ONPG assay is a single time point or end point measurement.
Another improvement is the exclusion of false positives caused by alkaline degradation of AHLs. The lactone bonds of AHLs can be rapidly hydrolyzed in an alkaline environment, resulting in ring opening. Previous research39 showed that the half-life of short-chain AHLs (C6, C7 and C8-HSL) was between 0.5 to 5.7 hours at high pH (8.2–9.55) and the shorter the acyl chain of AHLs the higher rate of the hydrolysis. Importantly, the ring-opened AHLs, which could also be caused by enzymolysis of AHL lactonase (e. g. AiiA13), could not activate signal receptor any more. In our initial tests, KH2PO4/K2HPO4 and Tris-HCl buffers (both with pH 6.7) were added to a final concentration of 100 mM when preparing Marine Broth 2216, but they were found unable to maintain slightly acid pHs effectively. The PIPES and the MOPS buffers were examined later and both of them were found to be effective to prevent alkaline degradation of C6-HSL. Additionally, the normalized β-galactosidase activities of the tested bacterial cultures did not show significant differences between the PIPES and the MOPS buffered groups. Although the MOPS buffer was successfully used by many researchers32,40, we finally chose the PIPES buffer for the A136 liquid X-gal assay because the pKa (where the maximum buffering capacity is achieved) of the PIPES is closer to the desired slightly acid pH (6.7) than the MOPS41. Additionally, the pH of a few bacterial cultures buffered by the MOPS became slighty alkaline during the following co-incubation but not in the PIPES buffered ones (data not shown) many false positives observed in our initial tests because of alkaline degradation were eventually removed after employing the PIPES buffer.
With the two major improvements, 25 QQ strains belonging to 14 species were successfully identified from 366 candidate bacterial strains and their QQ activities were either confirmed by the CV026 plate assay or by the VIR24 plate assay42. Romero et al43. identified 24 QQ strains out of 106 isolates from various marine environments with the CV026 plate assay and confirmed by HPLC-MS analysis, including strains with weak AHL degrading activities. Our assay could distinguish strong QQ activities from weak activities out of hundreds of isolates in a much more time-effective way.
The A136 liquid X-gal assay was successfully applied in identifying QQ bacteria among indigenous strains of healthy flounder and the QQ bacterial species obtained in this study showed great diversity (Table 1). Additionally, it was the first report of QQ activity in the genera Flaviramulus, Muricauda, Salinimonas, Thalassomonas, Marivita, Novosphingobium, Colwellia and Rhodobacter. Moreover, 12 out of 14 species (except the species of Olleya marilimosa44 and Tenacibaculum discolor43) were reported to bear QQ activities for the first time.
The C6-HSL degrading activities in three species were attributed to AHL lactonases (Table 1). Interestingly, the recovery of C12-HSL was dependent on the co-incubation time (Supplementary Fig. S4), which might be because the ring-opened C12-HSLs were further degraded by Flaviramulus spp. and Tenacibaculum soleae. QQ activities have been reported in some species of Tenacibaculum43,45 and the recovery of C6-HSL by acidification was only present in T. discolor 20J against C4-HSL but not in T. discolor 20J against 3-oxo-C12-HSL or T. maritimum NCIMB2154T against C10-HSL. Moreover, the recovery of C6-HSL could be observed in T. soleae (strains T133, T134, T148, T173, T189 and T195), but not in T. discolor T84 in our study. These results indicated that different types of QQ enzymes present in different species of Tenacibaculum or even in different strains of T. discolor. A complete recovery of C6-HSL was also observed in O. marilimosa T168, which showed strong degrading activities against all of the tested AHLs (Table 1) and this is in agreement with a recently reported QQ strain 138E in the same species44. However, the acidification assay of strain 138E against C12-HSL only showed a partial recovery of C12-HSL. This noteworthy difference to our observations indicated that there might be different enzymes (e.g. lactonase and acylase) existing in different strains of O. marilimosa.
Additionally, heat resistance of C6-HSL degrading activity was present in a relatively high percentage (4/12) of the QQ bacteria. Furthermore, the activities of Flaviramulus spp. and T. soleae strains were only partially abolished or not abolished at all, respectively, even when boiled for 30 min (Supplementary Fig. S5a online). Considering the lactonase activities in the two species, it was inferred that the lactonases of Flaviramulus spp. and T. soleae strains might be heat-resistant. Although several thermophilic QQ enzymes among a large number of QQ enzymes in various bacteria were reported46,47, it was still an exciting discovery suggesting that novel heat-resistant QQ enzymes may exist in these species. Further exploration of the QQ activities might unveil the diversity of AHL-degrading mechanisms.
AHL acylases might be more common than lactonases in marine QQ bacteria. Some QQ strains in our work only showed weak degrading activity against short-chain AHLs while strong activity against long-chain AHLs, or higher preference to C10 to C14-HSLs with or without a substitution of oxo-group (Figure 5 and Table 1). Moreover, Zobellia russellii T157 and Pseudoalteromonas paragorgicola Th100 also showed substrate specificity to C10 to C14-HSL without a substitution of oxo-group (Supplementary Fig. S5b online). These observations indicated that long-chain AHL degrading activities might be more common among our marine isolates, which is in agreement with the discoveries described previously43. AHL lactonases and acylases are two major types of QQ enzymes and lactonases normally presented broad AHLs inactivating activity while many acylases were specific to long-chain AHLs13,14. We assume that AHL acylases might be more common than lactonase in the ocean, which is in agreement with the distribution of acylase and lactonase sequences in metagenome collections44.
The application of QQ bacteria in aquaculture as an alternative to antibiotics to control disease have been proposed in several studies already10,11. Several positive effects on larval survival of brine shrimp and turbot were demonstrated upon the addition of microbial enrichment cultures with QQ activities17,18. However, the majority of the reported QQ bacteria are of terrestrial origin, which would limit the application in aquaculture30. Therefore, bacteria isolated from healthy flounder (gills, intestines and skin mucus) were chosen for screening QQ activities in our work and some QQ strains could be developed to probiotics to control disease in aquaculture. One of the mucus derived isolates was P. prydzensis Th125 and mussel related isolates in this species have recently been described to bear antibacterial activity against several vibrios48. Another mucus derived strain Muricauda olearia Th120 had cellular appendages and it is suggested that the appendages are required for connection of cells and adhesion to surfaces49. The epidermal mucus plays an important role in the fish innate immune system50 and the attachment to mucous surfaces was suggested as another criterion for probiotics51. Based on the properties of P. prydzensis Th125 and M. olearia Th120, it may be worthwhile to evaluate these two strains for their disease controlling potential in aquaculture.
Methods
Biosensor strains and growth conditions
The biosensor A. tumefaciens A136 (containing plasmids pCF218 and pCF372)21 lacks the Ti plasmid and provides AHL response system, the AHL-responsive transcription factor TraR (pCF218) and the TraR-regulated traI-lacZ fusion genes (pCF372). The biosensor A136 can detect a broad range of AHLs (C6 to C14-HSL). A136 was routinely maintained on Luria–Bertani (LB) agar supplemented with 50 μg ml−1 spectinomycin and 4.5 μg ml−1 tetracycline at 28°C and was grown in AT minimal medium52 (10.7 g KH2PO4, 2 g (NH4)2SO4, 160 mg MgSO4·7H2O, 10 mg CaCl2, 5 mg FeSO4·7H2O, 2 mg MnSO4·7H2O and 1 L distilled water; adjusted with 1 N KOH to pH 6.7) containing 0.5% (wt/vol) glucose for liquid X-gal assays. The biosensors CV02629 and VIR2442 are both cviI mutants derived from Chromobacterium violaceum ATCC 31532 and ATCC 12472T, respectively and produce purple pigment violacein in response to the short-chain (C4–C8) and long-chain (C8–C14) AHLs, respectively. CV026 and VIR24 were used to confirm the AHLs degrading activities of QQ strains identified by the A136 liquid X-gal assay and were routinely maintained on LB agar at 28°C.
N-acylhomoserine lactones (AHLs)
C6-HSL, 3-oxo-C6-HSL and C8-HSL were purchased from Cayman Chemical Company (Ann Arbor, Michigan, USA). 3-oxo-C8-HSL, C10-HSL, 3-oxo-C10-HSL, C12-HSL, 3-oxo-C12-HSL, C14-HSL and 3-oxo-C14-HSL were purchased from Sigma-Aldrich (St. Louis, Missouri, USA). All AHL stock solutions (10 mM) were prepared in DMSO.
Outline of the A136 liquid X-gal assay
The candidate bacterial strains were inoculated in Marine Broth 2216 (BD; Becton, Dickinson & Co.) and incubated at 28°C, 170 rpm for 24 h. Then the bacterial cultures were mixed with C6-HSL and the reaction was carried out at 28°C for 24 h. The supernatants of the reaction mixture were mixed with the A136 X-gal assay solution (an overnight broth culture of A136 inoculated in AT minimal glucose medium and mixed with X-gal to a final concentration of 250 μg ml−1) in 96-well plates and incubated at 28°C. The color development was monitored with Tecan Sunrise™ microplate absorbance reader at the wavelengths of 492 nm and 630 nm.
The absorbances at 492 nm and 630 nm are actually a combination of absorption and light scattering by indigo and A136 biosensor cells.




The correction factors a and b are constants.

Obtaining the correction factor values a and b for the normalization of β-galactosidase activity
In order to obtain the value of the correction factor a, an overnight broth culture of biosensor A136 was inoculated in AT minimal glucose medium. After incubation at 28°C for 24 h, the culture was serially diluted and the absorbances at wavelengths 630 nm and 492 nm were measured. When OD492 was plotted against OD630, the linear regression was analyzed and the slope was defined as the correction factor a.
In order to obtain the value of the correction factor b, an overnight broth culture of A136 was inoculated in AT minimal glucose medium and C6-HSL was added to a final concentration of 1 μM. After incubation at 28°C for 24 h, the culture was sonicated on ice. The supernatant of the culture was collected after centrifugation at 13 000 g for 10 min at 4°C and filtered through a 0.22 μm porosity filter. The filtrate was considered as the cell-free β-galactosidase solution. Then the cell-free β-galactosidase solution was mixed with X-gal (final concentration of 250 μg ml−1). After incubation at 28°C for 12 h, the blue product was serially diluted and the absorbances at 630 nm and 492 nm were measured. When OD630 was plotted against OD492, the linear regression was analyzed and the slope was defined as the correction factor b.
Optimization of the A136 liquid X-gal assay
Four factors in the reaction and the detection steps, including reaction buffer and pH, final concentration of C6-HSL (0.1 to 10 μM), inoculum size and incubation time, which may affect the accuracy of the A136 liquid X-gal assay for identifying QQ bacteria, were optimized. Briefly, the MOPS (pKa = 7.09) and PIPES (pKa = 6.71)41 buffers were prepared as stock solutions (1 M, pH 6.7) and were added to the bacterial culture to a final concentration of 100 mM. The pH of each reaction mixture was monitored.
In addition, an overnight broth culture (OD600 ≈ 2) of A136 was inoculated in AT minimal glucose medium and mixed with X-gal (final concentration of 250 μg ml−1). The resulting mixture was defined as the A136 X-gal assay solution. Different inoculum size (1%, 0.5% or 0.2%) of the A136 X-gal assay solution was tested and the normalized β-galactosidase activities were calculated every two hours as mentioned above. Three independent replicates were performed for each factor.
Comparison of the A136 liquid X-gal assay and the ONPG assay when monitoring the β-galactosidase activities
The optimized A136 liquid X-gal assay was compared with ONPG assay by monitoring the β-galactosidase activities of A136 induced by different concentrations of C6-HSL (0.1–0.7 μM), for verifying the feasibility, effectiveness and accuracy of the new method. The A136 liquid X-gal assay was carried out as above and the ONPG assay was carried out according to the protocol described by Miller24 with slight modifications. Briefly, the A136 X-gal assay solution without X-gal was incubated at 28°C for 12 h and the OD590 was measured. Then the cells of each sample were permeated with SDS and chloroform and then put in contact with ONPG. The β-galactosidase activities were monitored by OD415 and calculated by the formula:  (volume: the amount of A136 used at the beginning of the ONPG assay). Three independent replicates were performed for each assay.
(volume: the amount of A136 used at the beginning of the ONPG assay). Three independent replicates were performed for each assay.
Application of the A136 liquid X-gal assay for high-throughput identification of QQ bacteria
To verify the applicability of the A136 liquid X-gal assay, 366 bacterial strains, isolated from gills, mucus and intestines of healthy flounders from two fish farms in Shandong Province, China, were screened for QQ activities. The bacterial strains were cultured in Marine Broth 2216 at 28°C and their C6-HSL degrading activities were detected according to the final optimized protocol. The pHs of reaction mixtures were measured to ensure that there were no alkaline degradation of C6-HSL. The C6-HSL degrading activities of the positive strains were confirmed by the CV026 plate assay as described by McClean29.
Further characterization of AHL-degrading activities of QQ bacteria
The heat resistance of QQ activities was tested. Briefly, the bacterial cultures of QQ positive strains were boiled for 5 min and the levels of remaining C6-HSL of the reaction mixtures were measured by the A136 liquid X-gal assay and confirmed by the CV026 plate assay.
Additionally, lactonase activities of the QQ strains were demonstrated by acidification tests32. Briefly, after incubation for 24 h, the reaction mixture was boiled for 5 min and acidified with HCl to pH 2 followed by further incubation at 28°C for 24 h. The levels of remaining C6-HSL or C12-HSL in the reaction mixtures were measured by the A136 liquid X-gal assay and confirmed by the CV026 plate assay or VIR24 plate assay, respectively. The expressed AiiA protein of Bacillus thuringiensis in E. coli BL21(DE3)53 and P. aeruginosa ZJ551 (GenBank: FJ939361; isolated from seawater of the Yellow Sea Cold Water Mass) were used as controls for AHL lactonase and AHL acylase, respectively.
To evaluate the substrate range of the AHL-degrading activities of the QQ strains, AHLs with different acyl chains (3-oxo-C6-HSL, C8-HSL, 3-oxo-C8-HSL, C10-HSL, 3-oxo-C10-HSL, C12-HSL, 3-oxo-C12-HSL, C14-HSL and 3-oxo-C14-HSL) were used as substrates. The final concentration for the A136 liquid X-gal assay was 1 μM for C10-HSL, C12-HSL and C14-HSL and 100 nM for the rest of the AHLs. Furthermore, the degrading activities against these AHLs were confirmed by the VIR24 plate assay42, except for 3-oxo-C6-HSL, where the A136 plate assay was used.
16S rRNA gene-based bacterial identification and phylogenetic analysis
Bacterial strains were identified by 16S rRNA gene sequencing. Genomic DNAs were extracted and 16S rRNA genes were amplified using the universal primers B8F (5′-AGAGTTTGATCATGGCTCAG) and B1510 (5′-GGTTACCTTGTTACGACTT). PCRs were carried out under standard conditions43 and the 16S rRNA genes were sequenced and compared with those available in the EzTaxon Database54. 16S rRNA gene sequences were aligned using ClustalX55 with those isolates with known QQ activity30. Phylogenetic analysis was performed using the MEGA5 software package56 and trees were constructed using the neighbor-joining method and evaluated after 1000 bootstrap replicates.
Statistical analysis
The linear regression and correlation analysis were performed by GraphPad Prism software (version 5). The un-paired t-test was used to compare results.
Nucleotide sequence accession number
16S rRNA gene sequences of the 25 QQ bacterial strains in GenBank are under the accession numbers: JX412958, JX412957 and KC756863-KC756885.
References
Davies, D. G. et al. The involvement of cell-to-cell signals in the development of a bacterial biofilm. Science 280, 295–298 (1998).
Nealson, K. H., Platt, T. & Hastings, J. W. Cellular control of the synthesis and activity of the bacterial luminescent system. J. Bacteriol. 104, 313–322 (1970).
Greenberg, E. P., Hastings, J. W. & Ulitzur, S. Induction of luciferase synthesis in Beneckea harveyi by other marine bacteria. Arch. Microbiol. 120, 87–91 (1979).
Gambello, M. J. & Iglewski, B. H. Cloning and characterization of the Pseudomonas aeruginosalasR gene, a transcriptional activator of elastase expression. J. Bacteriol. 173, 3000–3009 (1991).
Krick, A. et al. A marine Mesorhizobium sp. produces structurally novel long-chain N-acyl-L-homoserine lactones. Appl. Environ. Microbiol. 73, 3587–3594 (2007).
Milton, D. L. et al. Quorum sensing in Vibrio anguillarum: characterization of the vanI/vanR locus and identification of the autoinducer N-(3-oxodecanoyl)-L-homoserine lactone. J. Bacteriol. 179, 3004–3012 (1997).
Milton, D. L. et al. The LuxM homologue VanM from Vibrio anguillarum directs the synthesis of N-(3-hydroxyhexanoyl)homoserine lactone and N-hexanoylhomoserine lactone. J. Bacteriol. 183, 3537–3547 (2001).
Schaefer, A. L., Hanzelka, B. L., Parsek, M. R. & Greenberg, E. P. Detection, purification and structural elucidation of the acylhomoserine lactone inducer of Vibrio fischeri luminescence and other related molecules. Methods Enzymol. 305, 288–301 (2000).
Croxatto, A. et al. VanT, a homologue of Vibrio harveyi LuxR, regulates serine, metalloprotease, pigment and biofilm production in Vibrio anguillarum. J. Bacteriol. 184, 1617–1629 (2002).
Finch, R. G., Pritchard, D. I., Bycroft, B. W., Williams, P. & Stewart, G. S. Quorum sensing: a novel target for anti-infective therapy. J. Antimicrob. Chemother. 42, 569–571 (1998).
Roy, V., Adams, B. L. & Bentley, W. E. Developing next generation antimicrobials by intercepting AI-2 mediated quorum sensing. Enzyme Microb. Technol. 49, 113–123 (2011).
Givskov, M. et al. Eukaryotic interference with homoserine lactone-mediated prokaryotic signalling. J. Bacteriol. 178, 6618–6622 (1996).
Dong, Y. H. et al. Quenching quorum-sensing-dependent bacterial infection by an N-acyl homoserine lactonase. Nature 411, 813–817 (2001).
Lin, Y. H. et al. Acyl-homoserine lactone acylase from Ralstonia strain XJ12B represents a novel and potent class of quorum-quenching enzymes. Mol. Microbiol. 47, 849–860 (2003).
Uroz, S. et al. N-Acylhomoserine lactone quorum-sensing molecules are modified and degraded by Rhodococcus erythropolis W2 by both amidolytic and novel oxidoreductase activities. Microbiology 151, 3313–3322 (2005).
Defoirdt, T. et al. Quorum sensing-disrupting brominated furanones protect the gnotobiotic brine shrimp Artemia franciscana from pathogenic Vibrio harveyi, Vibrio campbellii and Vibrio parahaemolyticus isolates. Appl. Environ. Microbiol. 72, 6419–6423 (2006).
Van Cam, D. T. et al. Novel approach of using homoserine lactone-degrading and poly-β-hydroxybutyrate-accumulating bacteria to protect Artemia from the pathogenic effects of Vibrio harveyi. Aquaculture 291, 23–30 (2009).
Tinh, N. T. N., Yen, V. H. N., Dierckens, K., Sorgeloos, P. & Bossier, P. An acyl homoserine lactone-degrading microbial community improves the survival of first-feeding turbot larvae (Scophthalmus maximus L.). Aquaculture 285, 56–62 (2008).
Nhan, D. T. et al. Quorum quenching bacteria protect Macrobrachium rosenbergii larvae from Vibrio harveyi infection. J. Appl. Microbiol. 109, 1007–1016 (2010).
Steindler, L. & Venturi, V. Detection of quorum-sensing N-acyl homoserine lactone signal molecules by bacterial biosensors. FEMS Microbiol. Lett. 266, 1–9 (2007).
Zhu, J. et al. Analogs of the autoinducer 3-oxooctanoyl-homoserine lactone strongly inhibit activity of the TraR protein of Agrobacterium tumefaciens. J. Bacteriol. 180, 5398–5405 (1998).
Zhu, J., Chai, Y., Zhong, Z., Li, S. & Winans, S. C. Agrobacterium bioassay strain for ultrasensitive detection of N-acylhomoserine lactone-type quorum-sensing molecules: detection of autoinducers in Mesorhizobium huakuii. Appl. Environ. Microbiol. 69, 6949–6953 (2003).
Mockli, N. & Auerbach, D. Quantitative β-galactosidase assay suitable for high-throughput applications in the yeast two-hybrid system. BioTechniques 36, 872–876 (2004).
Miller, J. H. Experiments In Molecular Genetics. (Cold Spring Harbor Laboratory Press, 1972).
Kawaguchi, T., Chen, Y. P., Norman, R. S. & Decho, A. W. Rapid screening of quorum-sensing signal N-acyl homoserine lactones by an in vitro cell-free assay. Appl. Environ. Microbiol. 74, 3667–3671 (2008).
Domingues, L., Teixeira, J. A. & Lima, N. Rapid and sensitive detection of β-galactosidase-producing yeasts by using microtiter plate assay. Biotechnol. Tech. 11, 399–402 (1997).
Khopkar, S. M. Basic Concepts Of Analytical Chemistry. (New Age International Publishers, 1998).
Spaun, J. Problems in standardization of turbidity determinations on bacterial suspensions. Bull. World Health Organ. 26, 219–225 (1962).
McClean, K. H. et al. Quorum sensing and Chromobacterium violaceum: exploitation of violacein production and inhibition for the detection of N-acylhomoserine lactones. Microbiology 143, 3703–3711 (1997).
Uroz, S., Dessaux, Y. & Oger, P. Quorum sensing and quorum quenching: the yin and yang of bacterial communication. ChemBioChem 10, 205–216 (2009).
d'Angelo-Picard, C., Faure, D., Penot, I. & Dessaux, Y. Diversity of N-acyl homoserine lactone-producing and -degrading bacteria in soil and tobacco rhizosphere. Environ. Microbiol. 7, 1796–1808 (2005).
Yates, E. A. et al. N-Acylhomoserine lactones undergo lactonolysis in a pH-, temperature- and acyl chain length-dependent manner during growth of Yersinia pseudotuberculosis and Pseudomonas aeruginosa. Infect. Immun. 70, 5635–5646 (2002).
Chan, K. G., Yin, W. F., Sam, C. K. & Koh, C. L. A novel medium for the isolation of N-acylhomoserine lactone-degrading bacteria. J. Ind. Microbiol. Biotechnol. 36, 247–251 (2009).
Christiaen, S. E., Brackman, G., Nelis, H. J. & Coenye, T. Isolation and identification of quorum quenching bacteria from environmental samples. J. Microbiol. Methods 87, 213–219 (2011).
McLean, R. J., Pierson, L. S., III & Fuqua, C. A simple screening protocol for the identification of quorum signal antagonists. J. Microbiol. Methods 58, 351–360 (2004).
Yang, Y. H. et al. High-throughput detection method of quorum-sensing molecules by colorimetry and its applications. Anal. Biochem. 356, 297–299 (2006).
Kim, Y. H., Kim, Y. H., Kim, J. S. & Park, S. Development of a sensitive bioassay method for quorum sensing inhibitor screening using a recombinant Agrobacterium tumefaciens. Biotechnol. Bioprocess Eng. 10, 322–328 (2005).
Singh, M. P. & Greenstein, M. A simple, rapid, sensitive method detecting homoserine lactone (HSL)-related compounds in microbial extracts. J. Microbiol. Methods 65, 32–37 (2006).
Decho, A. W. et al. Autoinducers extracted from microbial mats reveal a surprising diversity of N-acylhomoserine lactones (AHLs) and abundance changes that may relate to diel pH. Environ. Microbiol. 11, 409–420 (2009).
Chan, K. G. et al. Characterization of N-acylhomoserine lactone-degrading bacteria associated with the Zingiber officinale (ginger) rhizosphere: co-existence of quorum quenching and quorum sensing in Acinetobacter and Burkholderia. BMC Microbiol. 11, 51 (2011).
Fukada, H. & Takahashi, K. Enthalpy and heat capacity changes for the proton dissociation of various buffer components in 0.1 M potassium chloride. Proteins 33, 159–166 (1998).
Someya, N. et al. Distribution of N-acylhomoserine lactone-producing fluorescent pseudomonads in the phyllosphere and rhizosphere of Potato (Solanum tuberosum L.). Microbes Environ. 24, 305–314 (2009).
Romero, M., Martin-Cuadrado, A. B., Roca-Rivada, A., Cabello, A. M. & Otero, A. Quorum quenching in cultivable bacteria from dense marine coastal microbial communities. FEMS Microbiol. Ecol. 75, 205–217 (2011).
Romero, M., Martin-Cuadrado, A. B. & Otero, A. Determination of whether quorum quenching is a common activity in marine bacteria by analysis of cultivable bacteria and metagenomic sequences. Appl. Environ. Microbiol. 78, 6345–6348 (2012).
Romero, M., Avendaño-Herrera, R., Magariños, B., Cámara, M. & Otero, A. Acylhomoserine lactone production and degradation by the fish pathogen Tenacibaculum maritimum, a member of the Cytophaga–Flavobacterium–Bacteroides (CFB) group. FEMS Microbiol. Lett. 304, 131–139 (2010).
Seo, M. J., Lee, B. S., Pyun, Y. R. & Park, H. Isolation and characterization of N-acylhomoserine lactonase from the thermophilic bacterium, Geobacillus caldoxylosilyticus YS-8. Biosci. Biotechnol. Biochem. 75, 1789–1795 (2011).
Gotthard, G., Hiblot, J., Elias, M. & Chabriere, E. Crystallization and preliminary X-ray diffraction analysis of the hyperthermophilic Sulfolobus islandicus lactonase. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 67, 354–357 (2011).
Aranda, C. P. et al. Bacteriostatic anti-Vibrio parahaemolyticus activity of Pseudoalteromonas sp. strains DIT09, DIT44 and DIT46 isolated from Southern Chilean intertidal Perumytilus purpuratus. World J. Microbiol. Biotechnol. 28, 2365–2374 (2012).
Hwang, C. Y. et al. Muricauda olearia sp. nov., isolated from crude-oil-contaminated seawater and emended description of the genus Muricauda. Int. J. Syst. Evol. Microbiol. 59, 1856–1861 (2009).
Subramanian, S., MacKinnon, S. L. & Ross, N. W. A comparative study on innate immune parameters in the epidermal mucus of various fish species. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 148, 256–263 (2007).
Chabrillon, M., Rico, R. M., Balebona, M. C. & Morinigo, M. A. Adhesion to sole, Solea senegalensis Kaup, mucus of microorganisms isolated from farmed fish and their interaction with Photobacterium damselae subsp. piscicida. J. Fish Dis. 28, 229–237 (2005).
Tempe, J., Petit, A., Holsters, M., Montagu, M. & Schell, J. Thermosensitive step associated with transfer of the Ti plasmid during conjugation: possible relation to transformation in crown gall. Proc. Natl. Acad. Sci. U. S. A. 74, 2848–2849 (1977).
Bai, F., Han, Y., Chen, J. & Zhang, X.-H. Disruption of quorum sensing in Vibrio harveyi by the AiiA protein of Bacillus thuringiensis. Aquaculture 274, 36–40 (2008).
Kim, O. S. et al. Introducing EzTaxon-e: a prokaryotic 16S rRNA gene sequence database with phylotypes that represent uncultured species. Int. J. Syst. Evol. Microbiol. 62, 716–721 (2012).
Larkin, M. A. et al. Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 (2007).
Tamura, K. et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 (2011).
Huang, W. et al. QsdH, a novel AHL lactonase in the RND-type inner membrane of marine Pseudoalteromonas byunsanensis strain 1A01261. PLoS One 7, e46587 (2012).
Afriat, L., Roodveldt, C., Manco, G. & Tawfik, D. S. The latent promiscuity of newly identified microbial lactonases is linked to a recently diverged phosphotriesterase. Biochemistry 45, 13677–13686 (2006).
Acknowledgements
We thank Dr. Robert J.C. McLean (Texas State University, USA) for biosensors C. violaceum CV026 and A. tumefaciens A136 and Dr. Tomohiro Morohoshi (Utusnomiya University, Japan) for C. violaceum VIR24. This work was supported by the International Science and Technology Cooperation Programme of China (no. 2012DFG31990). T.C. and P.B. acknowledge support from FWO-Vlaanderen (no. G.A064.10N).
Author information
Authors and Affiliations
Contributions
X.-H.Z., K.T., T.C. and P.B. designed the experiments. K.T., Y.Z., M.Y. and X.S. performed the experiments. K.T. analysed the results. K.T., X.Z., T.C. and P.B. wrote the paper. All the authors offered a critical review of the paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Tang, K., Zhang, Y., Yu, M. et al. Evaluation of a new high-throughput method for identifying quorum quenching bacteria. Sci Rep 3, 2935 (2013). https://doi.org/10.1038/srep02935
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep02935
This article is cited by
-
Inhibitory effects of hexanal on acylated homoserine lactones (AHLs) production to disrupt biofilm formation and enzymes activity in Erwinia carotovora and Pseudomonas fluorescens
Journal of Food Science and Technology (2023)
-
Mechanisms and the role of probiotic Bacillus in mitigating fish pathogens in aquaculture
Fish Physiology and Biochemistry (2020)
-
Isolation and identification of pathogenic Vibrio spp. retrieved from diseased Litopenaeus vannamei and beneficial role of some functional probiotic bacteria for control
Aquaculture International (2020)
-
Prevention of biofilm formation by quorum quenching
Applied Microbiology and Biotechnology (2020)
-
The Quorum Quenching Bacterium Bacillus licheniformis T-1 Protects Zebrafish against Aeromonas hydrophila Infection
Probiotics and Antimicrobial Proteins (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
