
Abstract
A major drawback of traditional photocatalysts like TiO2 is that they can only work under illumination and the light has to be UV. As a solution for this limitation, visible-light-driven energy storage photocatalysts have been developed in recent years. However, energy storage photocatalysts that are full-sunlight-driven (UV-visible-NIR) and possess long-lasting energy storage ability are lacking. Here we report, a Pt-loaded and hydrogen-treated WO3 that exhibits a strong absorption at full-sunlight spectrum (300–1,000 nm) and with a super-long energy storage time of more than 300 h to have formaldehyde degraded in dark. In this new material system, the hydrogen treated WO3 functions as the light harvesting material and energy storage material simultaneously, while Pt mainly acts as the cocatalyst to have the energy storage effect displayed. The extraordinary full-spectrum absorption effect and long persistent energy storage ability make the material a potential solar-energy storage and an effective photocatalyst in practice.
Similar content being viewed by others

Introduction
The energy storage effect of photocatalysis materials is a phenomenon whereby photoinduced catalysis ability1, anticorrosion2, bactericidal effects3 or the reduction effect of poisonous heavy metal ions4 can last for a period of time even after the stoppage of the excitation, meaning that light energy absorbed by photocatalysis materials can be saved to release in dark. The study of energy storage materials has attracted increasing attentions in recent years, since it helps to overcome the drawback of photocatalysts like TiO2 that can only work under irradiation5. It has been rapidly developed in the past decade, largely stimulated by the TiO2−WO3 system with anticorrosion effect in dark2. To date, several energy storage material systems have been developed, such as TiO2-WO31,2,3,4,6,7,8,9, TiO2-MoO310, Au:TiO2-WO35, Au:TiO2-MoO35, SrTiO3-WO311, TiO2-Ni(OH)212,13 and TiO2-V2O514, which have been widely used in photocatalysis, anticorrosion and sterilization in dark.
In general, an energy storage material system is composed of two kinds of materials7,15: light harvesting materials and energy storage materials. Light harvesting materials are materials capable of absorbing light to generate electron-hole pairs. Energy storage materials are materials in charge of trapping and saving the electrons or holes transferred from light harvesting centers during illumination and releasing them in dark. Whereas the variety of the material system has been rapidly developed, the most frequently used light harvesting material is still TiO2. Thus, in most energy storage systems, UV light is used for illumination. To use the solar energy more efficiently, a few visible light harvesting materials5,15 have been investigated recently. However, the absorption of these materials in visible spectrum remains insufficient, due to the absorption threshold of them. What's more, even if the UV-visible light can be fully absorbed, the utilization of solar energy is still limited to 51%16. Thus full-spectrum light harvesting materials should be investigated, if utilization of solar energy to the full is to be realized. On the other hand, to overcome the drawback of photocatalysts that they can only function under irradiation, the energy storage time should be at least as long as 12 h, so that photocatalysts can not only be used during daytime, but also be used during the night. Although there have been persistent efforts to improve the energy storage time, few reports of long lasting energy storage effect have been proposed. Among the researched energy storage materials, WO3 is the favorite one, since it is the most viable and simple in construction of the hybrid materials and effective in storing the photogenerated electrons for long periods (over 12 h)17.
Thus, in the present, the most studied energy storage material system is still TiO2-WO3, in which TiO2 and WO3 act as light harvesting material and energy storage material, respectively. However, as mentioned above, the UV illumination limited the practical application of the TiO2-WO3 system. In fact, as a kind of semiconductor with the narrow band gap of ~2.6 eV, WO3 has a visible light induced self-photorecharge ability in a photoelectrochemical system15. Thus, WO3 itself can be used as both visible light harvesting material and energy storage material. Moreover, it is reported that hydrogen treatment can extend the absorption threshold of TiO2 to NIR, recently18.
Inspired from this, here we report that we have developed a Pt-loaded and hydrogen-treated WO3 that not only can be effectively activated by full-spectrum (UV-visible -NIR) light, but also has long persistent energy storage ability (over 300 h) to have formaldehyde degraded in dark. In this new material system, the hydrogen treated WO3 and the loaded Pt, functioning as the light harvesting-storage bi-functional material and the cocatalyst respectively, are both critical for the broad optical absorption and the energy storage performance of the material, endowing this new material potential applications in solar energy utilization and pollutant degradation in dark.
Results
The composition of this material is mainly substoichiometric WO3-x particles (particles sizes: 40–100 nm, Supplementary Fig. S1), of which the chemical formula can be written as W20O56 (Supplementary Fig. S2, S3). Pt nanoparticles are uniformly deposited on WO3-x particles (ca. 3 wt%, Supplementary Fig. S3, S4). In the preparation of the material, a two-step synthesis process was involved. Firstly, we conducted the hydrogen treatment through annealing commercial WO3 in hydrogen atmosphere. Then the hydrogen treated samples were loaded with Pt by an impregnation method. Details of the synthesis method are presented in the methods section. For the convenience of discussion, the material is denoted as Pt-H:WO3. Pt loaded only and hydrogen treated only WO3 are also prepared and denoted as Pt-WO3 and H:WO3, respectively. XRD and HRTEM results show that H:WO3 and Pt-H:WO3 consist of the same kind of substoichiometric WO3-x (Supplementary Fig. S2, S3), meanwhile, there is hardly any difference between the pristine WO3 and the Pt loaded WO3.
The hydrogen treatment induced an expanded optical absorption of Pt-H:WO3. Figure 1 shows the normalized UV-visible absorption spectra of the as-prepared samples at room temperature. All the samples show fairly identical absorbance in the UV region (<400 nm). At around 480 nm, the optical absorption of WO3 and Pt-WO3 decreases, consistent with the indirect band gap absorption edge of WO3 (~2.6 eV). Meanwhile, it's astonishing that the absorbance of H:WO3 and Pt-H:WO3 increases significantly from the wavelength of 480 nm up to 1000 nm, indicating a full-spectrum absorption. This is in agreement with the color changes from yellow to deep blue after the hydrogen treatment of WO3 (as shown in Fig. 1 inset). This phenomenon is different from the extension of optical absorption observed in the hydrogen-treated TiO2, in which the absorption in the visible and NIR region shows a sharp decrease18, suggesting this new material can utilize sunlight more efficiently.

UV–vis absorbance spectra of WO3, Pt-WO3, H:WO3 and Pt-H:WO3.
The inset shows the digital picture of the four samples.
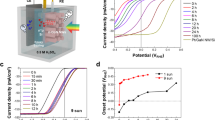
Besides the intense and broad absorption in the UV-visible and NIR spectrum, the Pt-H:WO3 sample also exhibits super-long-lasting energy storage ability after the removal of the excitation source. Figure 2a presents the formaldehyde degradation process in dark after being irradiated with a white light emitted from a flat-type LED for 1 h. From the degradation curves in Fig. 2a, it can be observed that WO3 and H:WO3 show no decomposition of formaldehyde at all, whereas Pt-WO3 and Pt-H:WO3 degraded formaldehyde both under irradiation and in dark. Furthermore, Pt-H:WO3 is well ahead of Pt-WO3 in degradation of formaldehyde in both situations, suggesting an enhanced degradation ability of Pt-H:WO3. These results were further confirmed by the evolved CO2 amounts shown in Fig. 2b.

Energy storage performances of the as-prepared four samples.
(a) Comparison of formaldehyde degradation ability of samples showing Ct/C0 versus time. The samples were irradiated with white light for 1 h at each beginning of the test. (b) Time courses of CO2 evolution during the formaldehyde degradation process. (c) Comparison of formaldehyde degradation ability in dark after being stored in dark for 12 h of Pt-H:WO3, Pt-WO3. (d) Gaseous-phase formaldehyde degradation in dark results of Pt-H:WO3, after being stored in dark for 36 h, 276 h, 300 h, recharged with white light for 1 h and being heated at 200°C for 2 h respectively.
To further evaluate the longtime energy storage ability of Pt-WO3 and Pt-H:WO3, we had the materials stored in dark for 12 h before the degradation test. As can be seen in Fig. 2c, whereas the comparable decomposition rate was shown in Fig. 2a just after the light irradiation than that in the light irradiation, after being stored in dark for 12 h, the degradation ability of Pt-WO3 decreased sharply, so as to less than 20% of formaldehyde was degraded at the end of the test. This suggests that the energy stored in Pt-WO3 should contribute to the decomposition of formaldehyde in dark and that the decrease of degradation efficiency of Pt-WO3 should be due to the limited energy storage ability of WO3. In contrast to Pt-WO3, after being stored in dark for 12 h, Pt-H:WO3 still maintained the high degradation ability and about 80% of formaldehyde was degraded in the end, meaning that the Pt-H:WO3 has a more durable energy storage ability with an energy storage time much longer than 12 h.
We prolonged the storage time in dark to further investigate the super-long persistent energy storage ability of Pt-H:WO3. Figure 2d presents the formaldehyde degradation performance of the same Pt-H:WO3 sample after being stored in dark for 36 h, 276 h and 300 h. As can be seen, when the storage time increased to 300 h, the sample still maintained the high degradation ability of formaldehyde, suggesting that the energy could be stored even longer than 300 h, a super-long energy storage time never reported before. To further demonstrate the long-lasting photocatalytic performance of Pt-H:WO3 photocatalyst coming from the storing energy of H:WO3 during light irradiation, the former tested sample was recharged with the white light for 1 h and then got tested in dark for the formaldehyde degradation performance (shown in Fig. 2d). It can be seen that after being recharged, the material recovered the formaldehyde degradation activity partially. It failed to recover to the initial degradation ability completely due to the accumulation of intermediate product, for after being heated at 200°C for 2 h, the material showed a completely recovered degradation activity, as shown in Fig. 2d. Thus, we conclude that the material gain the formaldehyde degradation ability from the excitation of light and can be recharged by light.
Discussion
The above results clearly show that the Pt-H:WO3 has a superb solar energy collection capability, persistent energy storage and high formaldehyde degradation efficiency in dark. To further elucidate the effect of the hydrogen treatment and how it interplays with the degradation activity, we measured the X-ray photoelectron spectroscopy (XPS) of all the samples, as shown in Fig. 3. The W 4f peaks can be decomposed into three pairs of peaks, corresponding to the typical binding energies of three W oxidation states, W6+, W5+and W4+ respectively, based on previous results19 (Fig. 3a). A similar work was done to O1s peaks to discriminate different oxygen species20,21 (Fig. 3b). The ratios of surface W6+/W5+/W4+ and O2−/OH−/H2O species are summarized in Supplementary Table S1. W5+ and W4+ found in the hydrogen-treated samples indicate the presence of oxygen vacancies, in agreement with the oxygen deficiency chemical formula gained from the XRD results (supplementary Fig. S2). Raman test results (Supplementary Fig. S5) further evidence that large amounts of oxygen vacancies were induced by the hydrogen treatment, in which almost all of the characteristic Raman peaks for monoclinic WO3 disappeared after hydrogen treatment, suggesting degradation of WO3 crystallinity, as expected for the increased amount of oxygen vacancies22. The disappearance of W4+ peak in Pt-H:WO3 should be ascribed to the weak surface oxidation caused by the sequent thermal treatment in air during Pt loading.

XPS investigation of WO3, H:WO3, Pt-WO3 and Pt-H:WO3.
(a) W 4f XPS spectra of as-prepared samples. The black curves correspond to the experimental data. Each black curve is decomposed into pairs of peaks corresponding to W6+ (blue curves), W5+(magenta curves) and W4+( dark yellow curves). (b) XPS O 1s core-level spectra of the four samples. The black curves correspond to the experimental data. Each black curve is decomposed into peaks corresponding to O2− (blue curve), OH− (cyan curve) and H2O (magenta curve).
In the normalized XPS O1s spectra of all the samples (Fig. 3b), we observed the lattice oxygen peak located at about 530 eV, hydroxyl (OH) peak at around 531.5 eV and a broader water (H2O) shoulder at around 532.5 eV. H:WO3 has the largest amount of OH groups, whereas the amount of surface OH groups of Pt-H:WO3 decreased and is fairly the same with that of Pt-WO3 and WO3, indicating that the hydroxyl groups induced by hydrogen treatment on the surface were removed during the sequent heat treatment in Pt loading procedure. The1H MAS NMR spectra further confirmed our hypothesis (supplementary Fig. S6), suggesting that the hydrogen treatment induced hydroxyl groups do not make a substantial contribution to the outstanding performance of Pt-H:WO3.
It's worth mentioning that during the fitting process of the profiles of hydrogen treated samples, we found that the peaks of the three W oxidation states all moved to the higher binding energy region, compared to the typical results19,22 and the pristine WO3 result. A similar tendency was observed in the fitting process of O1s peaks, with the consistent shift of about 0.5 eV. This phenomenon indicates a Fermi level shift caused by the super-high concentration of oxygen vacancies. It is well known that large amounts of lattice disorder in semiconductors could yield mid-gap states which often form a continuum extending to and overlapping with the conduction band (CB) edge, instead of forming discrete donor states near the CB edge. Such semiconductors are so-called degenerate semiconductors which generally show an upshift of the Fermi level. It is reported that in MoO3, similar in crystal and energy band structure with WO3, a certain degree of oxygen deficiency results in occupation of large density of states lying within the forbidden gap and CB with a corresponding shift of the Fermi level toward higher energies23.
These extended energy states in the band gap are also known as band tail states18, which usually become the dominant centers for optical excitation and relaxation. Broad absorption in UV-visible-NIR spectrum was observed in W18O49 and it was thought to be closely related to the oxygen deficiency24,25. Additionally, these extended energy states usually act as trapping sites for photogenerated carriers, preventing them from rapid recombination, thus promoting photocatalysis activity and energy storage effect. This is confirmed by PL spectra, which disclose the recombination process of photoinduced charge carriers. In general, the lower PL intensity indicates the lower recombination rate of photoinduced electron–hole pairs and thus the higher photocatalytic activity or energy storage ability of semiconductors26. According to the PL spectra (Fig. 4), the emission intensity of H:WO3, Pt-WO3 and Pt-H:WO3 samples is much lower than that of the original WO3, indicating a much repressed recombination rate and thus an efficient energy storage ability. Especially, the emission quenching of Pt-H:WO3 is such that it is hard to discern the peak, suggesting a very low recombination rate and an outstanding energy storage ability.

The PL spectra of as-prepared samples under excitation at 325 nm.
Thus, in our hydrogen treated samples, the oxygen vacancies, which are so dense to extend the defect band into the CB, form a degenerate semiconductor and shift Fermi level upward, should be responsible for the ultra-efficient absorption in UV-visible-NIR region and the super-long lasting energy storage ability.
In the PL spectra, the peak intensity shows in an order of WO3≫H:WO3>Pt-WO3>Pt-H:WO3, which is consistent with the formaldehyde degradation test result, except that H:WO3 shows no degradation of formaldehyde in our test. This suggests that H:WO3 also has an energy storage ability, but the energy stored can't be released through degradation of formaldehyde. This should be attributed to the more positive CB levels of H:WO3 than the reduction potentials of O2. It is generally considered that the CB level of a semiconductor should be more negative than the potential for the single-electron reduction of oxygen (O2 + e− = O2− (aq), −0.284 V vs NHE; O2 + H+ + e− = HO2 (aq), −0.046 V vs NHE) in order to allow efficient consumption of photoexcited electrons and subsequent oxidative decomposition of organic compounds by holes to proceed in air27. The CB levels of WO3 (+0.5 V vs NHE) is more positive than the reduction potentials of O2, thus WO3 is unsuitable for achieving the efficient oxidative decomposition of organic compounds in air. Thus H:WO3, having the same CB levels of WO3, shows a weak formaldehyde degradation ability because its CB level is not negative enough to facilitate the single-electron reduction of oxygen, even though electrons have been stored in the trapping sites.
On the other hand, Pt-H:WO3 and Pt-WO3 exhibit an enhanced formaldehyde degradation activity thanks to the promotion of multielectron reduction of O2 on the Pt cocatalyst. It has been reported that WO3 loaded with Pt nanoparticles exhibits high photocatalytic activity for the decomposition of organic compounds both in liquid and gas phases through promoting multielectron O2 reduction in a more positive potential on Pt which act as both an electron pool and cocatalyst27. Recently, it has been reported that when Pt is used as the cocatalyst with WO3, the excited electrons can react with O2 producing H2O2 and then producing OḢ radicals28, strong oxidant that can oxidize gaseous pollutant. This implies that Pt-loading can enhance the photocatalytic activity of WO3 and H:WO3 through facilitating both OḢ and holes decomposition of organic compounds by promoting multielectron O2 reduction. There are reports that suggest that Pt itself can catalyze the oxidation of formaldehyde into carbon dioxide and water29,30. However, our work suggests that the catalytic effect of metallic Pt alone on oxidation of formaldehyde should be very limited, even if the formaldehyde decomposition in dark of Pt-WO3 after being stored in dark for 12 h, shown in Fig.2c, is fully ascribed to the catalytic effect of metallic Pt. Thus, Pt loaded on WO3 and H:WO3, mainly functions as a co-catalyst to enhance the photocatalysis performance of WO3 and H:WO3 through providing the multielectron reduction of O2 pathway with a low overpotential.
In addition, Pt nanoparticles can act as an electron sink to capture, store and discharge electrons transferred from excited semiconductors31,32, allowing electrons to be discharged in dark. The stronger absorption of UV-visible-NIR light and the enhanced energy storage ability could enhance the photogenerated electrons transportation to Pt nanoparticles and prolong the discharge time of electrons in dark and thus promote and maintain the photocatalytic performance of Pt-H:WO3 in dark. Therefore, owing to a more effective harvest of sunlight and more intensive trapping sites induced by large amounts of oxygen vocancies, Pt-H:WO3 shows a higher formaldehyde degradation activity and a more persistent energy storage ability than those of Pt-WO3.
Considering all the test results above, we propose that the full spectrum and long persistent energy storage ability and high degradation efficiency of formaldehyde in dark of Pt-H:WO3 are acquired thanks to the enhanced charge activation, separation, storage owing to the enlarged defect band induced by hydrogen treatment and cocatalysis effect caused by Pt loading.
The mechanism is summarized as follows. The hydrogen treatment caused the formation of large amounts of oxygen vacancies in the tungsten trioxide (Fig. 5a), inducing a defect band in the band gap, which caused the upshift of the Fermi level (Fig. 5b) and the intensive full-spectrum absorption and provided trapping sites to store electrons for a super-long time (over 300 h). Pt nanoparticles loaded enhanced the formaldehyde degradation activity of the hydrogen treated WO3 through promoting the multielectron reduction of O2 on Pt.

A schematic representation of the full spectrum broad absorption and persistent energy storage mechanism of Pt-H:WO3.
(a) Reaction model of hydrogen treatment. (b) A schematic illustration of energy band structure of WO3, H:WO3, Pt-WO3, Pt-H:WO3. (c) The full-spectrum absorption and formaldehyde degradation in dark mechanism of Pt-H:WO3.
Under illumination, electrons activated transfer from the valence band (VB) to the conductive band or the defect band, or from the defect band to the conductive band (process 1, 3 and 4 in Fig. 5c), allowing absorption in UV-visible-NIR region. Meanwhile, trapping sites of the defect band also trap electrons to be stored, preventing electrons from recombination with holes in the VB (process 2). In dark, electrons stored in the defect band are released and transferred to Pt (process 5), to conduct the multielectron reduction of O2 (e.g., O2 + 2H+ + 2e− = H2O2(aq),+0.682 VNHE; O2 + 4H+ + 4e− = 2H2O,+1.23VNHE) (Fig. 5c process 6), thus facilitating the oxidation of formaldehyde, showing formaldehyde degradation activity in dark.
To summarize, we have developed a full-sunlight-driven photocatalyst with super-long persistent energy storage ability and enhanced formaldehyde degradation activity through hydrogen treating and Pt-loading of WO3. This material may find many important applications in solar energy utilization. For example, it can be used in solar cells, for it can be charged by light and saving electrons for a long time. And it can be used in the gas pollutant degradation both in the day and at night. Moreover, we have found that it is the hydrogen treatment generated oxygen vacancies that provide trapping sites to store electrons. Thus this study may open a new design strategy for developing energy storage material through simply adjusting the concentration of oxygen vacancies.
Methods
Materials synthesis
In the preparation of the material, a two-step synthesis process was involved. Firstly, we prepared the hydrogen treated samples through annealing commercial WO3 (of analytically pure grade, Tianjin kermel Chemical Reagent Co.Ltd., China) in hydrogen (1 bar, 5% H2, 95% Ar, 135 ml/min flow) in a tube furnace at 550°C for 2 h. Then the hydrogen treated samples were loaded with Pt by an impregnation method, in which WO3 powder (0.5 g) and distilled water (3 ml) containing chloroplatinic acid (0.0096 g/ml) were mixed in a beaker and left to be heated in a water bath (80°C). The suspension was evaporated to complete dryness under constant stirring. The resulting powder was calcined in air at 300°C for 2 h. A typical Pt loading on WO3 was estimated to be ca. 3 wt%.
Photocatalytic reactions
Photocatalytic degradation activities of the prepared samples under visible light were evaluated with a self-designed gas reactor. The total volume of this reactor is designed to be 1.25 L (The detailed descriptions of this reactor can be referred to our previous work33). The powder samples were spread out uniformly on a substrate made of aluminum. The samples was weighed to be 0.200 g for each test. The substrate could be installed in the gas reactor system. The whole area of the substrate was 50 × 50 mm2, which was irradiated by a flat-type white LED-light. The white light had a spectral range from 400 to 800 nm. The power of the light irradiated to the surface of photocatalytic sample was 5 mW/cm2. In our experiment, formaldehyde was chosen as a model pollutant in the experiment, which was a common indoor volatile harmful gas. The activities of the catalysts were evaluated in terms of the amount of formaldehyde degraded and CO2 generated during the photodegradation reactions. The analysis of carbon dioxide and formaldehyde concentration in the reactor was conducted on line with a Photoacoustic IR Multigas Monitor (Model 1412; INNOVA Air Tech Instruments). Prior to a test, gaseous formaldehyde gas (dry air and formaldehyde mixture) was allowed to reach adsorption and desorption equilibrium with the photocatalyst in the reactor. The initial concentration of formaldehyde was 100 ppm (±5 ppm) and that of water vapor was 1500 ppm (±50 ppm) which was 6.5% in relative humidity (RH). Each experiment lasted for 360 min at room temperature (about 20°C).
Physical characterization
UV-vis diffuse reflectance spectroscopy (DRS) absorption spectra of the samples were recorded at a UV-2550 spectrophotometer (PerkinElmer Lambda 35, Waltham, MA) using BaSO4 as the reference in the wavelength range of 200–1000 nm. Photoluminescence (PL) emission spectra were acquired under excitation at 325 nm using an USB2000-FLG Ocean Optics spectrometer. XPS data were collected using an X-ray photoelectron spectrometer Kratos XSAM800, employing Mg-Kα radiation. All the spectra were calibrated according to the binding energy of the C1s peak at 284.6 eV. Data were collected at room temperature.
References
Tatsuma, T., Saitoh, S., Ngaotrakanwiwat, P., Ohko, Y. & Fujishima, A. Energy storage of TiO2-WO3 photocatalysis systems in the gas phase. Langmuir 18, 7777–7779 (2002).
Tatsuma, T., Saitoh, S., Ohko, Y. & Fujishima, A. TiO2-WO3 photoelectrochemical anticorrosion system with an energy storage ability. Chemistry of Materials 13, 2838–2842 (2001).
Tatsuma, T., Takeda, S., Saitoh, S., Ohko, Y. & Fujishima, A. Bactericidal effect of an energy storage TiO2–WO3 photocatalyst in dark. Electrochemistry Communications 5, 793–796 (2003).
Zhao, D., Chen, C. C., Yu, C. L., Ma, W. H. & Zhao, J. C. Photoinduced Electron Storage in WO3/TiO2 Nanohybrid Material in the Presence of Oxygen and Postirradiated Reduction of Heavy Metal Ions. Journal of Physical Chemistry C 113, 13160–13165 (2009).
Takahashi, Y. & Tatsuma, T. Visible light-induced photocatalysts with reductive energy storage abilities. Electrochemistry Communications 10, 1404–1407 (2008).
Ngaotrakanwiwat, P. & Tatsuma, T. Optimization of energy storage TiO2–WO3 photocatalysts and further modification with phosphotungstic acid. Journal of Electroanalytical Chemistry 573, 263–269 (2004).
Cao, L., Yuan, J., Chen, M. & Shangguan, W. Photocatalytic energy storage ability of TiO2-WO3 composite prepared by wet-chemical technique. Journal of Environmental Sciences 22, 454–459 (2010).
Li, Y. J., Cao, T. P., Shao, C. L. & Wang, C. H. Preparation and Energy Stored Photocatalytic Properties of WO3/TiO2 Composite Fibers. Chemical Journal of Chinese Universities-Chinese 33, 1552–1558 (2012).
Ngaotrakanwiwat, P., Tatsuma, T., Saitoh, S., Ohko, Y. & Fujishima, A. Charge-discharge behavior of TiO2-WO3 photocatalysis systems with energy storage ability. Physical Chemistry Chemical Physics 5, 3234–3237 (2003).
Takahashi, Y., Ngaotrakanwiwat, P. & Tatsuma, T. Energy storage TiO2–MoO3 photocatalysts. Electrochimica Acta 49, 2025–2029 (2004).
Ohko, Y., Saitoh, S., Tatsuma, T. & Fujishima, A. SrTiO3-WO3 photocatalysis systems with an energy storage ability. Electrochemistry 70, 460–462 (2002).
Zhang, L. Y. et al. UV-Induced Oxidative Energy Storage Behavior of a Novel Nanostructured TiO2-Ni(OH)2 Bilayer System. Journal of Physical Chemistry C 115, 18027–18034 (2011).
Zhang, L. Y. et al. Enhanced energy storage of a UV-irradiated three-dimensional nanostructured TiO2-Ni(OH)2 composite film and its electrochemical discharge in the dark. Journal of Electroanalytical Chemistry 683, 55–61 (2012).
Ngaotrakanwiwat, P. & Meeyoo, V. TiO2-V2O5 Nanocomposites as Alternative Energy Storage Substances for Photocatalysts. Journal of Nanoscience and Nanotechnology 12, 828–833 (2012).
Ng, C., Ng, Y. H., Iwase, A. & Amal, R. Visible light-induced charge storage, on-demand release and self-photorechargeability of WO3 film. Physical Chemistry Chemical Physics 13, 13421–13426 (2011).
Casbeer, E., Sharma, V. K. & Li, X.-Z. Synthesis and photocatalytic activity of ferrites under visible light: A review. Separation and Purification Technology 87, 1–14 (2012).
Park, H., Bak, A., Jeon, T. H., Kim, S. & Choi, W. Photo-chargeable and dischargeable TiO2 and WO3 heterojunction electrodes. Applied Catalysis B-Environmental 115, 74–80 (2012).
Chen, X., Liu, L., Yu, P. Y. & Mao, S. S. Increasing solar absorption for photocatalysis with black hydrogenated titanium dioxide nanocrystals. Science 331, 746–750 (2011).
Leftheriotis, G., Papaefthimiou, S., Yianoulis, P. & Siokou, A. Effect of the tungsten oxidation states in the thermal coloration and bleaching of amorphous WO3 films. Thin Solid Films 384, 298–306 (2001).
Shpak, A. P., Korduban, A. M., Medvedskij, M. M. & Kandyba, V. O. XPS studies of active elements surface of gas sensors based on WO3−x nanoparticles. Journal of Electron Spectroscopy and Related Phenomena 156–158, 172–175 (2007).
Szilágyi, I. M. et al. WO3 photocatalysts: Influence of structure and composition. Journal of Catalysis 294, 119–127 (2012).
Wang, G. et al. Hydrogen-treated WO3 nanoflakes show enhanced photostability. Energy & Environmental Science 5, 6180–6187 (2012).
Vasilopoulou, M. et al. The Influence of Hydrogenation and Oxygen Vacancies on Molybdenum Oxides Work Function and Gap States for Application in Organic Optoelectronics. Journal of the American Chemical Society 134, 16178–16187 (2012).
Guo et al. Morphology-Controlled Synthesis of W18O49 Nanostructures and Their Near-Infrared Absorption Properties. Inorgannic Chemistry 51, 4763–4771 (2012).
Guo, Yin, Huang, Dong & Sato, T. Synthesis of W18O49 Nanorod via Ammonium Tungsten Oxide and Its Interesting Optical Properties. Langmuir 27, 12172–12178 (2011).
Liu, Y. et al. Improvement of gaseous pollutant photocatalysis with WO3/TiO2 heterojunctional-electrical layered system. Journal of Hazardous Materials 196, 52–58 (2011).
Abe, R., Takami, H., Murakami, N. & Ohtani, B. Pristine Simple Oxides as Visible Light Driven Photocatalysts: Highly Efficient Decomposition of Organic Compounds over Platinum-Loaded Tungsten Oxide. Journal of the American Chemical Society 130, 7780–7781 (2008).
Kim, J., Lee, C. W. & Choi, W. Platinized WO3 as an Environmental Photocatalyst that Generates OH Radicals under Visible Light. Environmental Science & Technology 44, 6849–6854 (2010).
Quiroz Torres, J., Royer, S., Bellat, J. P., Giraudon, J. M. & Lamonier, J. F. Formaldehyde: catalytic oxidation as a promising soft way of elimination. ChemSusChem 6, 578–592 (2013).
Zhang, C., He, H. & Tanaka, K.-i. Catalytic performance and mechanism of a Pt/TiO2 catalyst for the oxidation of formaldehyde at room temperature. Applied Catalysis B: Environmental 65, 37–43 (2006).
Takai, A. & Kamat, P. V. Capture, Store and Discharge. Shuttling Photogenerated Electrons across TiO2-Silver Interface. Acs Nano 5, 7369–7376 (2011).
Harris, C. & Kamat, P. V. Photocatalytic Events of CdSe Quantum Dots in Confined Media. Electrodic Behavior of Coupled Platinum Nanoparticles. Acs Nano 4, 7321–7330 (2010).
Zou, T. et al. Full mineralization of toluene by photocatalytic degradation with porous TiO2/SiC nanocomposite film. Journal of Alloys and Compounds 552, 504–510 (2013).
Acknowledgements
We thank the Nature Science Foundation of China (No. 50927201) and the National Basic Research Program of China (Grant Nos. 2009CB939705 and 2009CB939702) for financial support and Yao Yu, Lin Liu for assistance with Solid-state 1H MAS (magic angle spinning) NMR measurements. The authors are also grateful to Analytical and Testing Center of Huazhong University of Science and Technology.
Author information
Authors and Affiliations
Contributions
C.X., J.L. and Y.L. designed the experiments. C.X. was responsible for the project planning. J.L., Y.L., Z.Z(Zhu)., T.Z., Z.Z(Zou)., G.Z., S.Z., D.Z. performed experiments. J.L. and C.X. wrote the paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Li, J., Liu, Y., Zhu, Z. et al. A full-sunlight-driven photocatalyst with super long-persistent energy storage ability. Sci Rep 3, 2409 (2013). https://doi.org/10.1038/srep02409
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep02409
This article is cited by
-
Bacterial Polysaccharide-Stabilized Silver Nanoparticles Photocatalytically Decolorize Azo Dyes
Applied Biochemistry and Biotechnology (2023)
-
Electrochromism in Hf-doped WO3
Journal of Solid State Electrochemistry (2022)
-
Photocatalytic concrete for degrading organic dyes in water
Environmental Science and Pollution Research (2022)
-
Characterisation of oxygen defects and nitrogen impurities in TiO2 photocatalysts using variable-temperature X-ray powder diffraction
Nature Communications (2021)
-
Persistent CO2 photocatalysis for solar fuels in the dark
Nature Sustainability (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
