
Abstract
Mycobacterium tuberculosis modulates expression of various metabolism-related genes to adapt in the adverse host environment. The gene coding for M. tuberculosis S-adenosylhomocysteine hydrolase (Mtb-SahH) is essential for optimal growth and the protein product is involved in intermediary metabolism. However, the relevance of SahH in mycobacterial physiology is unknown. In this study, we analyze the role of Mtb-SahH in regulating homocysteine concentration in surrogate host Mycobacterium smegmatis. Mtb-SahH catalyzes reversible hydrolysis of S-adenosylhomocysteine to homocysteine and adenosine and we demonstrate that the conserved His363 residue is critical for bi-directional catalysis. Mtb-SahH is regulated by serine/threonine phosphorylation of multiple residues by M. tuberculosis PknB. Major phosphorylation events occur at contiguous residues Thr219, Thr220 and Thr221, which make pivotal contacts with cofactor NAD+. Consequently, phosphorylation negatively modulates affinity of enzyme towards NAD+ as well as SAH-synthesis. Thr219, Thr220 and Thr221 are essential for enzyme activity and therefore, responsible for SahH-mediated regulation of homocysteine.
Similar content being viewed by others

Introduction
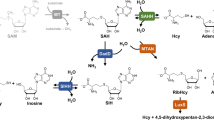
S-adenosylhomocysteine hydrolase (SahH) catalyzes reversible hydrolysis of S-adenosylhomocysteine (SAH) and produces homocysteine and adenosine1 (Fig. 1A). Homocysteine is either used in the transsulfuration pathway to synthesize cysteine or re-methylated to methionine for S-adenosylmethionine (SAM) regeneration2. Adenosine is either deaminated to inosine or used in the synthesis of nucleotides by salvage pathway2. Role of SahH in regulating the concentration of important metabolites such as SAH and homocysteine has been studied in great detail in eukaryotes3,4,5. In humans, genetic defect in SAHH gene (encoding SAHH/AdoHcy hydrolase) is accompanied by several impaired phenotypes including elevated levels of methionine and homocysteine3. SAH catabolism by SahH helps in increased tolerance to salt stress in plants by maintaining the synthesis of an osmolyte- glycine betaine4. Disruption of sahh in bacteria Rhodobacter capsulatus exhibits loss of survival in minimal media, altered SAM and SAH levels and decreased bacteriochlorophyll synthesis5. Similarly, SahH inhibitor neplanocin A increases SAH levels in Alcaligenes faecalis6. In Mycobacterium tuberculosis, the levels of SAH and homocysteine are modulated in response to different carbon sources and drugs respectively7,8. The mechanism underlying this regulation is still unknown. SahH may have an important role in the regulation of these metabolites in M. tuberculosis. sahh is essential for growth of M. tuberculosis9; however, the physiological role(s) of enzyme as well as its regulation is largely unexplored.

Metabolic pathways and activity of Mtb-SahH.
(A) Homocysteine metabolism and the role of SahH. SahH (green oval box) catalyzes reversible hydrolysis of SAH to homocysteine and adenosine as shown by bi-directional arrows. Numbers in circles denote the enzymes present in M. tuberculosis H37Rv as follows: 1- SahH (Rv3248c), 2- Cystathionine β-synthase (Rv1077), 3- Cystathionine γ-synthase (Rv1079), 4- Adenosine kinase (Rv2202c), 5- Adenylate kinase (Rv0733), 6- Adenosine deaminase (Rv3313c), 7- Methionine synthase (Rv2124c and Rv1133c), 8- S-adenosylmethionine synthetase (Rv1392) and 9- Methyltransferases, which transfer methyl group (CH3) to substrate molecules. (B) SAH-hydrolytic activity of His6-SahH purified from E. coli. 100 nM of holo-SahH was used with 0–100 μM of SAH. A reaction containing 100 μM SAH in the absence of enzyme (No enz.) served as a control for spontaneous SAH-hydrolysis. OD was measured at 415 nm and plotted as a function of time (minutes). SahH activity increased with increasing concentration of SAH. (C) SAH-synthetic activity of His6-SahH purified from E. coli. Reaction was performed for 2 hours using 4 μM of holo-SahH and TLC image was analyzed by autoradiography (lane 1). SahH from Rabbit erythrocytes was used as a positive control (lane 3) and a negative control was setup without the (lane 2). Adenosine (Ado) and SAH spots are indicated by arrows. After the densitometric analysis of spots, percent SAH synthesis is plotted in lower panel. An extra spot above SAH-spot in lane 3 could be inosine due to slight contamination (<1%) of adenosine deaminase enzyme in SahH (from rabbit erythrocytes purchased from a commercial source, Sigma) that deaminates adenosine to inosine. Corresponding retention factors (Rf) were SAH (0.1), inosine (0.36) and adenosine (0.5).
SahH consists of two domains- a substrate-binding catalytic domain and a cofactor binding domain10. The catalytic processes in both, SAH-hydrolytic and SAH-synthetic directions essentially require NAD+ as a cofactor11. M. tuberculosis is the first prokaryote in which the structure of SahH has been elucidated12. M. tuberculosis SahH (Mtb-SahH) forms a homotetramer with one NAD+ molecule bound to each subunit12. Residues involved in catalysis as well as those involved in hydrogen-bonding with NAD+ are conserved in Mtb-SahH. Interestingly, a distinct feature was observed near the homocysteine-binding site in solvent access channel involving His363 of Mtb-SahH12. In this study, we analyze the regulation of homocysteine concentration by Mtb-SahH. We show the role of the conserved residue His363 in enzyme activity. Subsequently, we study the regulation of SahH by serine/threonine phosphorylation, as well as identify important phospho-acceptor residues which, interestingly, are also involved in NAD+-binding. This study indicates that phosphorylation-mediated regulation of SahH is conserved in mycobacteria and it can be a novel mechanism of regulating homocysteine metabolism.
Results
Reversible catalysis of SAH by Mtb-SahH
To validate reversible catalysis, Mtb-SahH was cloned in pProEx-HTc and purified as a recombinant His6-tagged protein (HTc-SahH) from Escherichia coli. SAH-hydrolytic activity of purified Mtb-SahH was assessed by a colorimetric assay using Ellman's reagent (DTNB) that detects the product- homocysteine13. SahH requires NAD+ as a cofactor11 and its NADH-bound form is inactive14. Since the purified protein contains NAD+ as well as NADH14, there was no detectable activity in purified protein in the absence of any added cofactor (data not shown). Therefore, we prepared apo-SahH, reconstituted it with NAD+ to obtain holo-SahH, which was used with increasing concentration of SAH (0–100 μM) to analyze SAH-hydrolysis. An increase in SAH-hydrolytic activity was observed with increasing amounts of SAH (Fig. 1B), implying that holo-SahH was active. The reaction was found to attain saturation at 80 μM of SAH after which the enzyme activity did not increase further. The hydrolytic activity also increased with increasing amounts of holo-SahH (10–400 μM) (Supplementary Fig. S1). These results confirm SAH-hydrolytic activity of Mtb-SahH.
To validate that Mtb-SahH is also active in reversible direction, we analyzed its ability to synthesize SAH from homocysteine and adenosine. Mtb-SahH was incubated with [8-14C]-adenosine and homocysteine and the reactions were carried out for 1 or 2 hours. Reaction products were then resolved by thin layer chromatography (TLC) and analyzed by autoradiography. Apo-SahH showed negligible activity in the absence of NAD+ (Supplementary Fig. S2A), while holo-SahH could synthesize considerable amounts of SAH (Fig. 1C). Further, we observed that the activity increased with increasing concentration of holo-SahH (1–4 μM) (Supplementary Fig. S2B). Taken together, these assays show that M. tuberculosis SahH is an active enzyme that can catalyze reversible hydrolysis of SAH.
Role of His363 in SahH activity
SahH is one of the highly conserved proteins in both eukaryotes and prokaryotes10. His363 is a conserved residue in Mtb-SahH, which based on the sequence (Supplementary Fig. S3) and structural analyses, is proposed to act as a switch, that opens to allow access to the substrate SAH11,12. His363 needs to flip-out in order to accommodate homocysteine moiety of SAH12, although its role in enzyme activity has not been proven experimentally. To gain insight into this mechanism, His363 residue of Mtb-SahH was mutagenized to alanine and the SahH-H363A mutant was purified as a recombinant protein from E. coli. SAH-hydrolytic assays were performed with equal amounts of wild type SahH (SahH-WT) and SahH-H363A, where the mutant was found to be completely inactive (Fig. 2A). To confirm the role of His363 in catalysis we also checked the efficiency of H363A mutant to synthesize SAH. SahH-H363A mutant showed reduced activity to synthesize SAH, as compared to SahH-WT (Fig. 2B). These results confirm that His363 does act as a key residue for SahH enzyme activity.

Role of His363 in activity and effect of SahH on homocysteine concentration in M. smegmatis.
(A) Role of His363 in SAH-hydrolytic activity.Activities of 1.74 μM each of holo SahH-WT and holo SahH-H363A were compared. Graph shows increase in OD at 415 nm with time when SahH-WT was used. No increase in OD at 415 nm was observed with SahH-H363A mutant. Error bars represent standard deviation (SD). (B) Role of His363 in SAH-synthetic activity. Activities of 4 μM each of holo SahH-WT and holo SahH-H363A were compared. Autoradiograph of TLC shows SAH synthesis in reactions containing SahH-WT while reduced product formation was seen in reactions containing SahH-H363A. Adenosine (Ado) and SAH spots are indicated by arrows. (C) Histogram representing intracellular homocysteine concentration in M. smegmatis MC2 4517 with and without the over-expression of Mtb-SahH. Homocysteine concentration was analyzed using UPLC and plotted as μmoles/1015 CFU. Homocysteine levels increased to about six times in cells over-expressing SahH (see also Table 1). Error bars represent standard deviation of three independent experiments and * p < 0.05 as calculated by Student's t-test.
Regulation of homocysteine concentration by SahH
SahH catalyzes a reversible reaction where in vitro equilibrium favors the SAH-synthetic direction, but under in vivo conditions, hydrolytic activity is preferred as the products of this reaction (homocysteine and adenosine) are constantly being used by downstream enzymes1. In order to observe the favored direction of catalysis in mycobacteria, we examined the role of SahH in the regulation of intracellular homocysteine concentration in the surrogate host Mycobacterium smegmatis. Any change in the expression or activity of SahH may lead to an alteration in the concentration of homocysteine. Mtb-SahH was over-expressed in M. smegmatis MC2 4517 using pYUBDuet shuttle vector. Over-expression of Mtb-SahH was confirmed by immunoblotting using antibodies generated against Mtb-SahH (Supplementary Fig. S4). Total intracellular homocysteine concentration was then measured in M. smegmatis. The homocysteine amounts were 1.411 ± 0.25 μmoles/1015 colony forming units (CFU) in M. smegmatis (empty) and 1.346 ± 0.18 μmoles/1015 CFU in M. smegmatis harboring only the vector used for over-expression of Mtb-SahH (Table 1). Over-expression of Mtb-SahH increased homocysteine level to 6.363 ± 2.06 μmoles/1015 CFU (Table 1 and Fig. 2C). Thus, increase in levels of SahH in mycobacteria causes increase in this critical metabolite.
Conservation of SahH in mycobacteria and its regulation by phosphorylation
Since SahH is a key enzyme involved in metabolism, understanding its regulation will be helpful in revealing the mechanisms underlying homocysteine metabolism. In a prior study, during the analysis of intracellularly expressed proteins of M. tuberculosis, multiple spots of Mtb-SahH were found with distinct isoelectric points in two-dimensional electrophoresis, suggesting post-translational modifications of this protein15. Interestingly, Mtb-SahH was found to be a phosphorylated protein in a large-scale phosphoproteome analysis of M. tuberculosis16. Based on these earlier findings, we analyzed the phosphorylation status of SahH in different Mycobacterium species. SahH showed high amino acid sequence conservation in ten different species of Mycobacterium genus including both pathogenic and non-pathogenic bacteria (Supplementary Fig. S3). In order to assess conserved phosphorylation-mediated regulation of SahH across different mycobacterial species, we analyzed phosphorylation status of SahH in Mycobacterium bovis BCG and M. smegmatis. Native SahH from M. bovis BCG (Mbo-SahH) was immunoprecipitated and its phosphorylation status was analyzed. The identity of SahH in immunoprecipitated sample was first confirmed by immunoblotting with anti-SahH antibodies (Supplementary Fig. S5A). Phosphorylation was then confirmed by immunoblotting with anti-phospho-threonine (anti-pThr) antibodies as the major phosphorylated amino acid in M. tuberculosis is Thr16. Mbo-SahH was found to be phosphorylated (Fig. 3A). PknBc (catalytic domain of Mtb-PknB17, 1–331 amino acids) and GST were used as positive and negative controls, respectively. This result demonstrates that SahH is a potential substrate of serine/threonine protein kinases (STPKs) in M. bovis BCG.

In vivo phosphorylation of SahH and characterization of Msm-SahH.
(A) In vivo phosphorylation of SahH in M. bovis BCG.Native SahH was immunoprecipitated from M. bovis BCG and its phosphorylation was assessed by immunoblotting using anti-pThr antibodies. Upper panel shows immunoblot and lower panel shows ponceau-stained membrane. PknBc and GST were used as positive and negative controls, respectively. The protein band corresponding to SahH in Mbo-SahH was not visible on ponceau image but was found to be highly phosphorylated on Thr residues (See also Fig. S5A). (B) In vivo phosphorylation of SahH in M. smegmatis. Mtb-SahH was cloned and over-expressed in M. smegmatis as a His6-tagged protein and phosphorylation was assessed in purified fraction (pSD5-SahH) by immunoblotting using anti-pThr antibodies. Upper panel shows immunoblot and lower panel shows ponceau-stained membrane. PknBc and PknBc-K40M were used as positive and negative controls, respectively (See also Fig. S5B). (C) Detection of Msm-SahH by immunoblotting. Msm-SahH was over-expressed and purified from E. coli and analyzed by immunoblotting using anti-SahH antibodies. Upper panel shows immunoblot and lower panel shows ponceau-stained membrane. Mtb-SahH and GST were used as positive and negative control respectively. (D) Activity analysis of Msm-SahH purified from E. coli as a His6-tagged protein. 1.74 μM of holo Msm-SahH was used for the activity analysis and OD at 415 nm is plotted as a function of time. Graph shows increase in SAH-hydrolytic activity over time.
Further, phosphorylation of Mtb-SahH was confirmed in M. smegmatis. Mtb-SahH was over-expressed as a His6-tagged protein and purified from M. smegmatis. Purified fraction (pSD5-SahH) was phospho-enriched and immunoblotted with anti-SahH and anti-pThr antibodies. The phospho-enriched sample contained multiple protein bands and presence of SahH was confirmed by immunoblotting with anti-SahH antibodies (Supplementary Fig. S5B). Protein identity was also confirmed by mass spectrometry (data not shown). Immunoblotting with anti-pThr antibodies showed that Mtb-SahH is phosphorylated in M. smegmatis by native STPKs (Fig. 3B). PknBc and PknBc-K40M18 (catalytically inactive mutant of PknB) were used as positive and negative controls, respectively. These results together confirm that SahH gets phosphorylated in vivo in different species such as M. bovis BCG and M. smegmatis suggesting that SahH is a conserved substrate of STPKs.
M. smegmatis sahh was initially thought to be a pseudogene with a frame-shift mutation, although later it was corrected and proved to be a sequencing error19, still there was no conclusive experimental proof of presence of active M. smegmatis SahH (Msm-SahH). We therefore, cloned the gene coding for Msm-SahH in pProEx-HTc and purified the protein from E. coli. In sequence analysis, this gene did not show any frame-shift mutation and the sequence coded an ORF of 485 amino acids (data not shown). Anti-SahH antibodies generated against Mtb-SahH were able to detect the purified Msm-SahH (Fig. 3C). Also, Msm-SahH was found to be enzymatically active (Fig. 3D) confirming conservation of SahH in M. smegmatis.
In vitro phosphorylation and dephosphorylation of Mtb-SahH
Ser/Thr phosphorylation is a vital component of signaling machinery of M. tuberculosis consisting of 11 STPKs (PknA to PknL) and one Ser/Thr phosphatase (PstP)20. Out of 11 STPKs, only four (PknA, PknB, PknG and PknL) are conserved in Mycobacterium leprae, which has evolved after extensive gene decay and elimination, leaving only the genes that are critical for survival21. pknA and pknB (the genes coding for PknA and PknB, respectively) are located in the same genomic region as pstP (the gene coding for PstP)20. The gene coding for PknB is essential for the growth and survival of M. tuberculosis22 and PknB regulates several fundamental processes like cell shape, cell division, metabolism, protein synthesis and response to stress18,23,24,25,26,27. To assess the phosphorylation of SahH by PknB, in vitro kinase assays were performed with Mtb-SahH and PknBc. As seen in the autoradiograph (Fig. 4A), Mtb-SahH was found to be phosphorylated by PknBc. Phosphorylation was specifically localized to the protein and not on the His6-tag as evident after the cleavage of tag by TEV protease (Supplementary Fig. S6). We also found that SahH is a target of multiple STPKs of M. tuberculosis (data not shown), which is consistent with earlier observation16. The reversible regulation of Mtb-SahH by phosphorylation was confirmed by the dephosphorylation assay with PstPc (catalytic domain of PstP28, 1–300 amino acids). SahH phosphorylated by PknBc was found to be dephosphorylated by PstPc in a time-dependent manner (Fig. 4B). These experiments show that SahH is a substrate of Mtb-STPK PknB and Ser/Thr phosphatase PstP.

Phosphorylation of Mtb-SahH by PknBc.
(A) In vitro kinase assay of Mtb-SahH with PknBc. Upper panel shows autoradiograph with PknBc alone (lane 1) and PknBc with SahH (lane 2). Lower panel shows the corresponding coomassie-stained gel image. As evident, SahH is phosphorylated by PknBc. (B) In vitro dephosphorylation of Mtb-SahH by PstPc. SahH phosphorylated by PknBc was subjected to dephosphorylation by PstPc and samples were taken for analysis at the indicated time points, for a total of 25 minutes. Upper panel shows autoradiograph and lower panel shows coomassie-stained gel image. (C) Autoradiograph of metabolic labeling of SahH-P and SahH-UP. E. coli expressing SahH-P or SahH-UP was subjected to metabolic labeling and autoradiograph is shown. SahH co-expressed with PknB (SahH-P) was observed to be phosphorylated as opposed to SahH co-expressed with PknB-K40M (SahH-UP). (D) Phosphoamino acid analysis of SahH-P by 2D-TLE. Left panel shows ninhydrin-stained TLC plate and right panel shows the corresponding autoradiograph. Residues pThr, pSer and pTyr have been encircled. As observed in the autoradiograph, only pThr is detected in SahH phosphorylated by PknB. (E) Immunoblot analysis with anti-pThr antibodies. SahH-P and SahH-UP were purified and their phosphorylation status was analyzed. Upper panel shows the immunoblot and lower panel shows ponceau-stained membrane. SahH-P was found to be phosphorylated on Thr residues.
Phosphorylation of Mtb-SahH in the surrogate host E. coli
To further substantiate PknB-specific in vitro SahH phosphorylation, Mtb-SahH was co-expressed in E. coli with PknB in a dual expression vector pETDuet-1. As a control, catalytically inactive mutant PknB-K40M18 was co-expressed with Mtb-SahH instead of PknB. To discern the in vivo phosphorylation of SahH, E. coli cells containing the dual expression constructs were metabolically labeled with 32P-orthophosphoric acid to label the phosphorylated proteins. Ni+-NTA affinity pull-down was then used to extract SahH and purified proteins were run on SDS-PAGE followed by autoradiography. pETDuet-SahH:PknB (renamed SahH-P) was found to be phosphorylated while pETDuet-SahH:PknB-K40M (renamed SahH-UP) was not phosphorylated (Fig. 4C). To identify the phosphorylated amino acid residues of SahH, phosphoamino acid analysis was performed with purified SahH-P followed by two-dimensional thin layer electrophoresis (2D-TLE). Phosphorylation was present specifically on Thr residues (pThr) while neither phospho-Ser (pSer) nor phospho-Tyr (pTyr) was detected on SahH-phosphorylated by PknB in E. coli (Fig. 4D). To validate Thr phosphorylation, SahH-P and SahH-UP were purified as His6-tag proteins and analyzed for the phosphorylation by immunoblotting using anti-pThr antibodies after their interaction with PknB or PknB-K40M in E. coli cells. While, SahH-UP was found to be unphosphorylated, SahH-P was found to be phosphorylated on Thr residues (Fig. 4E). These experiments reaffirm that Mtb-SahH is phosphorylated by PknB.
Identification and validation of phosphorylation sites
Purified SahH-P and SahH-UP were subjected to mass-spectrometric analysis to identify the phosphorylation sites. Five Thr and one Ser residues were found to be phosphorylated on SahH by PknB in E.coli (Fig. 5A). Among the phospho-sites, Thr216, Thr219, Thr220 and Thr221 are present in the domain I, which is the substrate binding catalytic domain. Thr257 and Ser376 are present in the domain II which is the cofactor binding domain12. No phosphorylated residues were identified on SahH-UP (data not shown).

Analysis of phosphorylation sites on Mtb-SahH.
(A) Diagrammatic representation of phosphorylation sites at SahH-P identified by mass-spectrometry. Five Thr (Thr216, Thr219, Thr220, Thr221 and Thr257) and one Ser residues (Ser376) were identified and labeled above the respective domains of SahH. Domain I is shown in blue color and domain II is shown in white color. (B) and (C) Histograms depicting relative phosphorylation of SahH and its single or multiple Thr mutants, respectively. Purified SahH-WT and its mutants were analyzed by immunoblotting with anti-pThr antibodies. Percent relative phosphorylation was calculated considering the phosphorylation intensity of SahH-WT as 100%. Corresponding immunoblots are shown in Supplementary Fig. S7A and S7B. (D) Metabolic labeling of SahH-WT and SahH-T219A/T220A/T221A mutant. Histogram depicting densitometric analysis of autoradiograph of metabolically labeled proteins purified from E. coli. Percent relative phosphorylation is shown considering phosphorylation intensity of SahH-WT as 100%. Corresponding autoradiograph and coomassie-stained gel image is shown in Supplementary Fig. S8.
To validate these phosphorylation sites of SahH, specific Ser and Thr residues were mutated to alanine to generate phospho-deficient variants of SahH which cannot accept a phosphate group. SahH-WT and mutant proteins co-expressed with PknB in E. coli were thus purified, analyzed for phosphorylation by immunoblotting and subsequent densitometric analysis was plotted. Single site mutants of Thr219, Thr220, Thr221 and Thr257 residues showed 25–50% loss in phosphorylation signal except SahH-T216A which showed hyper-phosphorylation (Fig. 5B, Supplementary Fig. S7A). No considerable loss in phosphorylation was observed in SahH-S376A by immunoblotting using anti-pSer antibodies (data not shown). Multiple Thr mutants of SahH were then generated and analyzed in the same way. Maximum loss (approximately 90%) in the phosphorylation signal was obtained in the triple mutant of Thr219, Thr220 and Thr221 (SahH-T219A/T220A/T221A) (Fig. 5C, Supplementary Fig. S7B). To substantiate the phosphorylation of these three residues, E. coli containing SahH-WT or SahH-T219A/T220A/T221A, both co-expressed with PknB, were subjected to metabolic labeling as discussed in previous section and proteins were analyzed by autoradiography. The SahH-T219A/T220A/T221A mutant showed approximately 60% loss in phosphorylation intensity when compared to SahH-WT (Fig. 5D, Supplementary Fig. S8). Thus, Thr219, Thr220 and Thr221 are the major sites of phosphorylation at SahH by PknB. A mutant of all the four Thr residues at sites 216, 219, 220 and 221 (SahH-T216A/T219A/T220A/T221A) was also generated and found to be hyperphosphorylated (Supplementary Fig. S7B). The mutation of Thr at the position 216 could have resulted in structural perturbations leading to opening of tertiary structure, which served as a non-specific substrate for the phosphorylation. As a result, both SahH-T216A and SahH-T216A/T219A/T220A/T221A mutants were hyperphosphorylated (Fig. 5B and Supplementary Fig. S7B).
Phosphorylation-mediated changes in SahH activity
The major phospho-sites at SahH (Thr219–Thr221) obtained in our study with PknB are also involved in NAD+ binding12. Therefore, we chose to analyze the phosphorylation-mediated changes in the affinity of SahH towards NAD+. To assess the kinetic differences in the activity of SahH due to its phosphorylation, SAH-hydrolytic activities were measured with the apo-forms of SahH-P and SahH-UP using increasing concentration of NAD+. Phosphorylation of Mtb-SahH by PknB in E. coli did not affect the Vmax of SAH-hydrolysis although the Km for cofactor NAD+ increased significantly (Fig. 6A). While the Km of unphosphorylated SahH was 0.31 ± 0.042 μM, phosphorylation significantly increased it to 1.08 ± 0.122 μM (Table 2). Thus, phosphorylation decreased the affinity of SahH for NAD+, confirming that the Thr219–Thr221 residues play a decisive role in NAD+ binding. Similar negative effect of phosphorylation was observed when the enzyme activity of SahH was assessed in synthetic direction. SahH-P was less efficient in the synthesis of SAH using adenosine and homocysteine (Fig. 6B). Densitometric analysis indicated that SahH-P was only 50% efficient in the synthesis of SAH as compared to SahH-UP (Fig. 6C).

Effect of phosphorylation on enzyme activity of Mtb-SahH.
(A) Effect of phosphorylation on SAH-hydrolytic activity. Michaelis-Menten plot of SahH-P and SahH-UP is shown with concentration of NAD+ at horizontal axis. Phosphorylation of SahH is accompanied by increase in Km and no change in Vmax (see also Table 2). (B) Effect of phosphorylation on SAH-synthetic activity. Autoradiograph of TLC is shown with reactions containing 4 μM each of SahH-P, SahH-UP and no enzyme. Intensity of SAH spot decreases when SahH-P is used as compared to SahH-UP. (C) Histogram showing percent SAH synthesis by SahH-P and SahH-UP as observed in (B). SahH-P is only 50% active in synthesis of SAH. Error bars represent SD of three independent results. *** p value equal to 0.001 as calculated by Student's t-test. (D) Role of Thr219, Thr220 and Thr221 in SAH-hydrolytic activity. Activities of 1.74 μM each of holo SahH-WT (SahH-P) and holo SahH-T219A/T220A/T221A (marked as SahHtri) purified from E. coli were compared. Graph shows increase in OD at 415 nm of SahH-P with time while SahHtri does not show any detectable activity.
Since the major sites of phosphorylation were found to be Thr219–Thr221, we analyzed the enzyme activity of SahH-T219A/T220A/T221A mutant. We hypothesized that if the side-chains of these three Thr residues are involved in NAD+ binding, then the mutant should not be able to bind the cofactor. We found that SahH-T219A/T220A/T221A mutant had no SAH-hydrolytic activity when compared to SahH-WT protein (Fig. 6D), supporting our hypothesis. These results show the importance of Thr219–Thr221 residues and their phosphorylation in regulating SahH activity.
Structural analysis of NAD+ binding
To further understand the phosphorylation-mediated changes in the cofactor binding, we analyzed the available crystal structure of Mtb-SahH (PDB ID: 2ZIZ) with respect to the NAD+ binding. NAD+ binds to the domain II of SahH (Fig. 7A) but still makes close contacts with the three Thr residues at positions 219, 220 and 221 of domain I (Fig. 7B). As shown in the figure, the side-chain oxygen atoms (OG1) of all the three Thr residues were within 5 Å reach of the oxygen atom (O2D) at the C-2′ of the ribose moiety containing the nicotinamide of NAD+. The region encompassing the Thr219, Thr220 and Thr221 residues of SahH was then enlarged to show their interaction with NAD+ (Fig. 7C). We next used UCSF Chimera to simulate the presence of pThr at the positions 219, 220 and 221. Phosphorylation imparts two negative charges to the side-chain of each Thr by adding a phosphate group containing three oxygen atoms (Fig. 7D). This charge possibly disrupts the electrostatic interaction of the protein with NAD+. Although NAD+ may still bind to SahH, the efficiency with which it binds may greatly diminish. Also, to accommodate the phosphate group, additional structural perturbation may occur leading to interference with NAD+ binding. This analysis explains the possible reasons for the decrease in affinity of SahH-P for NAD+ and loss in activity of SahH-T219A/T220A/T221A mutant.

Structural analysis of SahH and its major phosphorylation sites.
(A) Cartoon representation of Mtb-SahH bound to NAD+ cofactor. Both the domains of SahH have been labeled. Thr219–Thr221 residues have been colored (magenta) to mark their proximity with NAD+ (purple). Other color representations are: helix (cyan), strand (yellow) and turn (red). (B) Region depicting interaction of NAD+ (green) with Thr219–Thr221. Side chains of Thr residues are shown in orange color. Distances of side-chain oxygen of Thr residues from oxygen of NAD+ are: 2.855 Å (Thr219), 4.239 Å (Thr220) and 3.155 Å (Thr221). Other color representations are according to that in (A). (C) and (D) Stick diagram representation of Thr219–Thr221 residues with NAD+. Side-chain oxygen molecules of Thr residues were modified to attach a phosphate group with two negative charges in (D). Color representations are as follows: Thr219–Thr221 (red), NAD+ (green), Phosphorus (yellow) and Oxygen (white).
Validation of SahH phosphorylation sites in M. smegmatis
To further validate the role of Thr219–Thr221 residues in SahH phosphorylation, we mutated these residues simultaneously in pYUBDuet-SahH and over-expressed SahH-T219A/T220A/T221A in M. smegmatis MC2 4517. Over-expression was confirmed by immunoblotting using anti-SahH antibodies (Supplementary Fig. S4). We used two-dimensional gel electrophoresis to resolve the isoforms of SahH after the mutation of three contiguous Thr residues. After this, the different isoforms of SahH were analyzed by immunoblotting using anti-SahH antibodies. We identified four isoforms of SahH-WT, separated according to their isoelectric points (Fig. 8A). Interestingly, one isoform was lost when SahH-T219A/T220A/T221A mutant was used indicating loss of phosphorylation. This shows that the Thr219–Thr221 residues are important phosphorylation sites of Mtb-SahH in M. smegmatis.

Validation of Thr219–Thr221 as phosphorylation sites and role in homocysteine regulation.
(A) Validation of Thr219–Thr221 as phosphorylation sites in M. smegmatis. SahH-WT and SahH-T219A/T220A/T221A were purified from M. smegmatis, separated by two-dimensional PAGE and immunoblotted with anti-SahH antibodies. For clarity only the region containing SahH isoforms has been shown and different isoforms have been encircled. As seen, one isoform is lost in SahH-T219A/T220A/T221A. (B) Role of Thr219–Thr221 residues in regulating homocysteine concentration. Histogram shows intracellular homocysteine concentration in M. smegmatis MC2 4517 with and without the over-expression of SahH-T219A/T220A/T221A (marked as SahHtri). Homocysteine concentration was analyzed using UPLC and plotted as μmoles/1015 CFU. Homocysteine levels were found to be unaffected by SahH-T219A/T220A/T221A (see also Table 1). Error bars represent SD of three independent results.
Importance of Thr219–Thr221 in regulating homocysteine levels
We next used SahH-T219A/T220A/T221A as a phospho-deficient as well as enzymatically inefficient derivative to study the role of SahH in regulating homocysteine concentration. Homocysteine amounts were measured in M. smegmatis MC2 4517 with over-expression of SahH-T219A/T220A/T221A. No measurable alteration in homocysteine levels were observed in cells over-expressing SahH-T219A/T220A/T221A mutant as compared to vector-alone (Table 1 and Fig. 8B), while SahH-WT increased homocysteine concentration (Table 1 and Fig. 2C). These results validate the role of Thr219–Thr221 residues in SahH-mediated regulation of homocysteine concentration in vivo.
Discussion
Homocysteine is a key intermediate in the synthesis of metabolites such as methionine, cysteine, SAM, SAH and adenosine (Fig. 1A). Sulfur-containing metabolites- methionine and cysteine are required for the protein synthesis while SAM is essential for the one-carbon metabolism. In M. tuberculosis, transsulfuration pathway helps in conversion of methionine to cysteine where homocysteine is an intermediate29. Adenosine is metabolized via the purine salvage pathway and proteins involved in this pathway are considered to be drug targets in M. tuberculosis30. Thus, homocysteine stands at an important metabolic junction where it helps in regulating the levels of crucial metabolites. Although the genes required in these metabolic reactions have been identified in M. tuberculosis, there is no information on regulation of the homocysteine metabolism. Here, we present the evidence for the role of SahH in homocysteine metabolism, its reversible enzyme mechanism and role of phosphorylation in its regulation.
Our first objective was to characterize reversible enzyme activity of Mtb-SahH. NAD+ was found to be a requisite for both SAH-hydrolytic and SAH-synthetic activities. NAD+-bound SahH was active and did not require any other cofactor or metal ion. Further, loss in SAH-hydrolytic and decrease in SAH-synthetic activity by H363A mutation strengthens the earlier structural observation, where His363 needs to flip out in order to accommodate ethylthioadenosine (ETA, a partial analog of SAH)12. His363 is thus a critical residue for enzyme activity.
We further analyzed functional conservation of SahH by measuring its effect on homocysteine concentration. In earlier studies, attempts to measure homocysteine-thiolactone in absence of exogenous homocysteine supplements could detect only very low levels in M. smegmatis31. Even using mass spectrometry based techniques, the sensitivity of detecting endogenous homocysteine was very low and it is only the upregulation that was reported in the presence of drug8. In our study, we used Ultra-high performance liquid chromatography (UPLC) to separate the thiol-containing molecules in combination with a sensitive method of fluorescence-based detection. We were able to detect homocysteine in concentrated cell-free extracts of M. smegmatis. We found that concentration of homocysteine in M. smegmatis was indeed very low (Table 1) and over-expression of SahH increases homocysteine levels to about six times. This increase was in accordance with our in vitro results where SAH hydrolysis augmented when SahH amounts were increased. Over-expression of SahH in M. smegmatis led to increased SAH-hydrolysis resulting in elevated homocysteine levels. This also indicates that the probable favorable direction of catalysis in vivo is SAH-hydrolysis; however, further investigations are required to confirm this hypothesis. SahH may regulate concentration of metabolites like homocysteine, adenosine and SAH in the cell and any change in its activity may perturb their concentration in the cell. As discussed in later sections, our study suggests that SahH activity can be regulated by phosphorylation of critical residues involved in NAD+ binding.
We investigated the phosphorylation-mediated regulation of SahH using M. tuberculosis PknB as a representative of the M. tuberculosis STPKs. We observed that Mtb-SahH is a substrate of PknB among other kinases. Since reversible regulation is a defining characteristic of STPK-mediated phosphorylation, we checked dephosphorylation of SahH using PstPc. SahH was found to be dephosphorylated by PstPc in a time-dependent manner. Phosphorylation of SahH was also evaluated in the E. coli dual expression system co-expressing SahH and PknB. A convincing way to look at in vivo phosphorylation is metabolic labeling using 32P-orthophosphoric acid. In this experiment SahH-P (phosphorylated by PknB) was phosphorylated while SahH-UP (co-expressed with PknB-K40M) was unphosphorylated. Phosphoamino acid analysis helped us to identify the phospho-residues in SahH. In this analysis, Thr residues were found to be phosphorylated. SahH was then purified (SahH-P and SahH-UP) from E. coli where SahH-P was found to be phosphorylated at Thr residues while SahH-UP was unphosphorylated. These experiments confirmed that SahH is modified by reversible Ser/Thr phosphorylation mediated by PknB and PstP.
Mass spectrometry analysis revealed that PknB phosphorylates SahH on multiple Ser/Thr residues. Interestingly, three Thr residues (Thr219, Thr220 and Thr221) were also identified by Prisic et al. in M. tuberculosis phosphoproteome16. This indicates that PknB is likely to be the kinase responsible for SahH phosphorylation in M. tuberculosis. Also, four different isoforms of Mtb-SahH were identified when it is purified from M. smegmatis. This suggests that M. smegmatis STPKs phosphorylate Mtb-SahH on multiple residues reaffirming our mass spectrometry results. Multiple phosphorylation events at a single protein are a common phenomenon in M. tuberculosis signaling. In M. tuberculosis phosphoproteome 301 phosphoproteins were identified with 516 sites, implying that there are several proteins that are phosphorylated on multiple sites16. The multiple sites of phosphorylation individually contributed almost equally to total phosphorylation at SahH as loss in any single residue does not completely abrogate the phosphorylation intensity. But, when multiple Thr residues were mutated to alanine, a triple mutant SahH-T219A/T220A/T221A showed maximum loss in phosphorylation. Importance of these three Thr residues in phosphorylation was also seen in vivo by metabolic labeling of E. coli expressing SahH-T219A/T220A/T221A and PknB. Here again, SahH-T219A/T220A/T221A mutant showed significant loss in phosphorylation.
SahH was found to be a conserved protein with high sequence identity among mycobacterial species and the conservation of Thr219–Thr221 residues is noticeable. In light of this observation, we reveal that phosphorylation of SahH is also a conserved phenomenon in Mycobacterium species. Native SahH from M. bovis BCG was found to be phosphorylated. Also, recombinant Mtb-SahH was found to be phosphorylated in M. smegmatis suggesting a common phosphorylation-mediated regulatory mechanism of SahH. Since Thr219–Thr221 residues were proposed to be involved in NAD+ binding12, we suspected that multiple phosphorylated residues on SahH may affect its enzymatic activity. Therefore, we measured kinetic parameters of SahH-P and SahH-UP in SAH-hydrolytic direction. Phosphorylation accompanied increase in Km indicating decrease in affinity towards NAD+, the essential cofactor of SahH. Since NAD+ is required for activity in both the directions, phosphorylated SahH was found to be only 50% efficient in SAH-synthesis as compared to unphosphorylated SahH. Loss in activity of SahH-T219A/T220A/T221A mutant reinforced the role of Thr219–Thr221 residues in NAD+ binding. NAD+ binds in a pocket constituted by domain II, which is a Rossmann fold, but also makes electrostatic interactions with residues in domain I12. We analyzed this interaction and observed that the distance between side chains of Thr219–Thr221 residues and ribose oxygen of NAD+ is ≤ 5 Å. Phosphorylation of Thr219–Thr221 modifies its interaction with NAD+ and thus modulates activity of SahH. We also simulated the replacement of Thr219–Thr221 residues with pThr by modifying the atoms in already available structure of SahH. It was clear that there would be hindrance in the binding of NAD+ once the residues at these positions are phosphorylated. Interestingly, Thr219–Thr221 residues are also important for SahH-mediated regulation of homocysteine concentration as SahH-T219A/T220A/T221A mutant does not upregulate homocysteine concentration in M. smegmatis.
In E. coli, acetate has been proposed to inhibit re-methylation of homocysteine to methionine leading to elevated intracellular homocysteine concentration which is toxic to the cell33,34. Although such inhibitory effect of homocysteine needs to be elucidated in mycobacteria, our results indicate that exogenous homocysteine retards the growth of M. smegmatis (data not shown). Phosphorylation of SahH may be used in certain conditions to lower its activity and consequently prevent upsurge of homocysteine concentration. Interestingly, SahH phosphorylation was found to be induced in the presence of acetate16. However, further investigations are required to assess the role of SahH and its phosphorylation in homocysteine-mediated toxicity.
Loss of NAD+ binding either by mutation of critical amino acid residues (Thr219–Thr221) or by phosphorylation leads to decreased SahH activity and the latter mechanism may be used by mycobacteria to fine tune the homocysteine concentration in the cell. Recently, another report had shown that phosphorylation of M. tuberculosis SahH by PknA affects Vmax of SAH-hydrolytic activity32. Thr2 and Thr221 were identified as phosphorylation sites but the mechanism by which phosphorylation at these sites decreases Vmax remains obscure. Here we not only reveal the effect of phosphorylation on SahH activity but also provide the evidence that phosphorylation affects NAD+ binding and thus has an overall negative effect on catalysis. We find a novel regulator of homocysteine concentration in mycobacteria and suggest a mechanism that governs its modulation. SahH, a highly conserved protein, is regulated by phosphorylation and the phenomenon is conserved across Mycobacterium species. The regulation of SahH by phosphorylation is an important phenomenon as it governs concentration of key metabolites.
Methods
Bacterial strains and gene manipulations
E. coli cells were grown and maintained in LB medium as described35. M. smegmatis MC2 155, M. smegmatis MC2 451736 (kindly provided by Dr. Rajesh S. Gokhale) and M. bovis BCG were grown in Middlebrook 7H9 broth supplemented with 0.5% glycerol, 10% ADC, 0.05% Tween-80. The gene coding for SahH (rv3248c and msmeg_1843) was PCR amplified from M. tuberculosis H37Rv and M. smegmatis MC2 155 genomic DNA, respectively and cloned in pProEx-HTc, pETDuet-1 (MCS-1), pSD537 and pYUBDuet38 vectors. Gene coding for PknB or PknB-K40M (full-length, 1–626 aa) was inserted in MCS-2 of pETDuet-127,39. Cloning of PknBc, PknBc-K40M and PstPc in pProEx-HTc was performed as explained earlier40. pETDuet-SahH or pYUBDuet-SahH was used as template to generate mutations using QuikChange XL-site directed mutagenesis kit, according to manufacturer's instructions (Stratagene). Tables 3 and 4 provide the detailed information about plasmid constructs and primers used throughout this study.
Protein expression and purification
For expression of His6-tagged proteins from pProEx-HTc or pETDuet-1, BL21 (DE3) cells were transformed with recombinant plasmids. Proteins were expressed and purified as described earlier40. The purified proteins were analyzed by SDS-PAGE and concentrations were estimated by Bradford assay (Bio-Rad).
Enzymatic assay of SahH
E. coli-purified His6-SahH and its mutants were subjected to ammonium sulfate precipitation to remove bound nucleotides, using the protocol of Gomi et al.41. Detailed protocol is described in Supplementary methods. The ‘apo’ forms of protein thus obtained were free of NAD+ cofactor. To reconstitute the active ‘holo’ form, enzymes were incubated with 1 mM β-NAD+ for 30 minutes at 4°C in buffer A (50 mM potassium phosphate buffer [pH 7.0] and 1 mM EDTA).
Enzyme activity was measured in hydrolytic direction using a colorimetric assay13 with 0–100 μM SAH. Reaction buffer contained 100 μM Ellman's reagent (or DTNB, Sigma) and 1 unit of adenosine deaminase (Sigma) in buffer A. Reaction was started by adding 100 nM of holo-form of SahH, unless otherwise indicated. For the determination of Km and Vmax, 1.74 μM of apo-forms of SahH-P and SahH-UP were used with 0–10 μM β–NAD+. The reaction was allowed for 25 min at 37°C and OD was measured at 415 nm (microplate reader 680 XR, Bio-Rad). Absorbance units were converted to activity units (pmoles/mg/min). Km and Vmax values were obtained by non-linear regression analysis using GraphPad Prism software. Values from three independent experiments were presented as mean ± standard error.
Enzymatic activity in synthetic direction was measured as described42. Briefly, 1–4 μM of holo-SahH was incubated with 25 μCi [8-14C] adenosine (American Radiolabeled Chemicals, St. Louis, MO; specific activity 53.5 mCi/mmol) and 5 mM DL-homocysteine (Sigma) in 20 mM potassium phosphate buffer [pH 7.0] (with 1 mM EDTA and 1 mM DTT). Reaction samples were incubated at 37°C for one and two hours followed by boiling at 100°C for 5 minutes. 10 μl sample from the total reaction volume of 50 μl was spotted on cellulose thin-layer chromatography (TLC) plates (Merck) and developed with butanol:methanol:water:NH4OH (60:20:20:1, v/v). Plates were dried and analyzed by autoradiography using Personal Molecular Imager (PMI, Bio-Rad). Densitometric analysis was done using Quantity One® 1-D analysis software (Bio-Rad). Percent SAH synthesis was calculated as fraction of substrate adenosine converted to SAH.
Homocysteine measurement
M. smegmatis MC2 4517 cells were grown to OD600 of 0.6–0.8 and induced with 1 mM IPTG and 0.2% acetamide for 48 hours. Cells were harvested and washed three times with sterile water. Harvested cells were dissolved in sterile water and lysed by bead beating using 0.1 mm Zirconium beads. Cell debris was removed by centrifugation and cell-free lysate was processed for homocysteine measurement as described43,44. Briefly, 100 μl of cell lysate was treated with 35 μl of 1.43 M Sodium borohydride dissolved in 0.1 N NaOH (to reduce the disulfide bonds). Then, 10 μl of n-amyl alcohol was added followed by addition of 35 μl of 1 N HCl. To this, 50 μl of 7 mM bromobimane (B4380, Sigma) dissolved in 5 mM sodium EDTA [pH 7.0] was added (to conjugate the reduced thiols with fluorophore). The solution was incubated at 42°C for 12–15 minutes followed by incubation at room temperature for 30–45 minutes. 50 μl of 1.5 M perchloric acid was used to precipitate proteins. Supernatant was neutralized with 6 μl of 2 M tris base. 4 μl of derivatized sample was injected into a 100 × 4.6 mm, 1.8 micron Eclipse plus C18 column using Agilent-1260 Ultra-high performance liquid chromatography (UPLC). Column was equilibrated with 90% buffer 1 (5% methanol and 0.86% acetic acid) and 10% Buffer 2 (100% methanol). Thiols were eluted from the column with linear gradient of both buffers (from 90% Buffer 1:10% Buffer 2 to 0% Buffer 1:100% buffer 2 in 7 minutes), with flow rate of 0.7 ml/min. Standard curve was generated with known concentrations of homocysteine (0.195–3.125 μM). Results were quantified by taking the area for the homocysteine-bromobimane peak and calculating its concentration using a regression equation derived from the standard curve. Homocysteine concentration was represented as μmoles/1015 CFU and results from three independent experiments were plotted as mean ± standard deviation.
Generation of polyclonal antibodies for SahH in mouse and immunoblotting
Affinity purified His6-SahH was used for antibody generation (see Supplementary methods). Standard protocols for western blotting were followed as described earlier40. The antibodies and dilutions used were- anti-pThr antibody (Monoclonal, Cell Signaling) 1:20,000 dilution, anti-pSer antibody (Abcam) 1:20,000 dilution, anti-SahH antibody 1:20,000 dilution and HRP-conjugated anti-mouse IgG antibody (Bangalore Genei) 1:20,000 dilution. The blots were developed using Immobilon™ western chemiluminescent HRP substrate kit (Millipore) according to manufacturer's instructions. During analysis of Thr mutants for loss in phosphorylation, densitometric analysis of bands was done using alphaimager 3400 (Alpha Innotech Corporation, San Leandro, California, USA) and normalized to their corresponding ponceau images. The relative loss in phosphorylation was calculated by considering the phosphorylation of SahH-WT as 100%.
Immunoprecipitation of SahH
To assess the phosphorylation status of native SahH, M. bovis BCG was grown till the OD600 = 1. Cells grown in 10 ml media were harvested and resuspended in 1X PBS containing protease inhibitor cocktail followed by lysis using bead-beating. Native SahH was immunoprecipitated from 2 mg whole cell lysate using anti-SahH antibodies following the protocol as described by the manufacturer (Pierce® Crosslink Immunoprecipitation Kit). The immunoprecipitated sample was run on SDS-PAGE, analyzed by immunoblotting using anti-pThr and anti-SahH antibodies.
Purification of SahH from M. smegmatis
For purification of His6-SahH from M. smegmatis electroporated with pSD5-SahH, similar protocol was followed as described earlier27. The purified protein was phosphoenriched, concentrated and utilized for immunoblotting. Identity of SahH was also confirmed by mass-spectrometric analysis (TCGA, New Delhi). For purification of pYUBDuet-SahH, plasmid constructs were electroporated in M. smegmatis MC2 4517 and transformants were selected on 7H10 agar plates containing Kan and Hyg. Purification procedures were the same as with pSD5-SahH except that the cultures were induced with 1 mM IPTG.
In vitro kinase assays and phosphatase assays
In vitro phosphorylation of 4–5 μg SahH by PknBc (1–2 μg) was carried out as described45 using 2 μCi [γ-32P] ATP. For the visualization of phosphorylation signal on cleaved proteins, removal of recombinant tag was achieved by a previous protocol27. In vitro dephosphorylation assays were carried out by adding PstPc (1 μg) as described by Sajid et al.46. In all the assays proteins were separated by 10–12% SDS-PAGE and analyzed by PMI.
Metabolic labeling in E. coli
E. coli (BL21-DE3) transformants harboring either pETDuet-SahH:PknB, pETDuet-SahH:PknB-K40M or pETDuet-SahH-T219A/T220A/T221A:PknB were used for metabolic labeling as described by Sajid et al46. Extracted samples were analyzed by autoradiography using PMI. Densitometric analysis of autoradiograph-signals was done using alphaimager 3400 and normalized to amount of protein as seen in their respective coomassie-stained images.
Phosphoamino acid analysis
For phosphoamino acid analysis, metabolically labeled SahH-P was used and similar protocol was followed as previously described45,47. Detailed protocol is described in Supplementary methods.
Identification of phosphorylation sites at SahH-P
For identification of phosphorylation sites, SahH-P and SahH-UP (3 μg) was run on 12% SDS-PAGE, stained with coomassie, de-stained and band corresponding to SahH-P was excised from the gel and washed with MilliQ water. The samples were processed for identification of phosphorylation sites by using Thermo-Finnagen LTQ electrospray instrument (Proteomics Core Facility, Children's Hospital, Boston, USA). Detailed sample processing protocol has been described earlier46.
Sequence and structural analysis of proteins
Amino acid sequence analysis of SahH was done using clustalW248, using ten different species of Mycobacterium. Available structure of Mtb-SahH was downloaded from PDB website (PDB ID: 2ZIZ) and analyzed in UCSF chimera12,49. Atoms were labeled using 2D labels in chimera tools and Phosphate group was added by modifying atoms using chimera tools.
Analysis of proteins by two-dimensional PAGE
Recombinant proteins purified from M. smegmatis MC2 4517 were desalted using chloroform/methanol precipitation and 1 μg of both the proteins were resolved by two-dimensional PAGE as described previously27. Isoelectric focusing was done using 7 cm IPG strips (Bio-Rad) with 4–7 pH range, followed by second dimensional SDS-PAGE. After this, proteins were transferred to nitrocellulose membrane and immunoblotted using anti-SahH antibodies.
References
De La Haba, G. & Cantoni, G. L. The enzymatic synthesis of S-adenosyl-L-homocysteine from adenosine and homocysteine. J. Biol. Chem. 234, 603–608 (1959).
Finkelstein, J. D. Regulation of homocysteine metabolism. In: Homocysteine in Health and Disease (eds Carmel, R., Jacobsen, D.) P 92–99 (Cambridge University Press, Cambridge, U.K. 2001).
Baric, I. et al. S-adenosylhomocysteine hydrolase deficiency in a human: a genetic disorder of methionine metabolism. Proc. Natl. Acad. Sci. U. S. A. 101, 4234–4239 (2004).
Weretilnyk, E. A. et al. Maintaining methylation activities during salt stress. The involvement of adenosine kinase. Plant Physiol. 125, 856–865 (2001).
Sganga, M. W., Aksamit, R. R., Cantoni, G. L. & Bauer, C. E. Mutational and nucleotide sequence analysis of S-adenosyl-L-homocysteine hydrolase from Rhodobacter capsulatus. Proc. Natl. Acad. Sci. U. S. A. 89, 6328–6332 (1992).
Fisher, E. W., Decedue, C. J., Keller, B. T. & Borchardt, R. T. Neplanocin A inhibition of S-adenosylhomocysteine hydrolase in Alcaligenes faecalis has no effect on growth of the microorganism. J. Antibiot. (Tokyo) 40, 873–881 (1987).
Griffin, J. E. et al. Cholesterol catabolism by Mycobacterium tuberculosis requires transcriptional and metabolic adaptations. Chem. Biol. 19, 218–227 (2012).
Chakraborty, S., Gruber, T., Barry, C. E., III, Boshoff, H. I. & Rhee, K. Y. Para-aminosalicylic acid acts as an alternative substrate of folate metabolism in Mycobacterium tuberculosis. Science 339, 88–91 (2013).
Sassetti, C. M., Boyd, D. H. & Rubin, E. J. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48, 77–84 (2003).
Turner, M. A. et al. Structure and function of S-adenosylhomocysteine hydrolase. Cell Biochem. Biophys. 33, 101–125 (2000).
Palmer, J. L. & Abeles, R. H. The mechanism of action of S-adenosylhomocysteinase. J. Biol. Chem. 254, 1217–1226 (1979).
Reddy, M. C. et al. Crystal structures of Mycobacterium tuberculosis S-adenosyl-L-homocysteine hydrolase in ternary complex with substrate and inhibitors. Protein Sci. 17, 2134–2144 (2008).
Lozada-Ramirez, J. D., Martinez-Martinez, I., Sanchez-Ferrer, A. & Garcia-Carmona, F. A colorimetric assay for S-adenosylhomocysteine hydrolase. J. Biochem. Biophys. Methods 67, 131–140 (2006).
Matuszewska, B. & Borchardt, R. T. The role of nicotinamide adenine dinucleotide in the inhibition of bovine liver S-adenosylhomocysteine hydrolase by neplanocin A. J. Biol. Chem. 262, 265–268 (1987).
Singhal, N., Sharma, P., Kumar, M., Joshi, B. & Bisht, D. Analysis of intracellular expressed proteins of Mycobacterium tuberculosis clinical isolates. Proteome Sci. 10, 14 (2012).
Prisic, S. et al. Extensive phosphorylation with overlapping specificity by Mycobacterium tuberculosis serine/threonine protein kinases. Proc. Natl. Acad. Sci. U. S. A. 107, 7521–7526 (2010).
Boitel, B. et al. PknB kinase activity is regulated by phosphorylation in two Thr residues and dephosphorylation by PstP, the cognate phospho-Ser/Thr phosphatase, in Mycobacterium tuberculosis. Mol. Microbiol. 49, 1493–1508 (2003).
Kang, C. M. et al. The Mycobacterium tuberculosis serine/threonine kinases PknA and PknB: substrate identification and regulation of cell shape. Genes Dev. 19, 1692–1704 (2005).
Deshayes, C. et al. Interrupted coding sequences in Mycobacterium smegmatis: authentic mutations or sequencing errors? Genome Biol. 8, R20 (2007).
Cole, S. T. et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544 (1998).
Cole, S. T. et al. Massive gene decay in the leprosy bacillus. Nature 409, 1007–1011 (2001).
Fernandez, P. et al. The Ser/Thr protein kinase PknB is essential for sustaining mycobacterial growth. J. Bacteriol. 188, 7778–7784 (2006).
Park, S. T., Kang, C. M. & Husson, R. N. Regulation of the SigH stress response regulon by an essential protein kinase in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 105, 13105–13110 (2008).
Parikh, A., Verma, S. K., Khan, S., Prakash, B. & Nandicoori, V. K. PknB-mediated phosphorylation of a novel substrate, N-acetylglucosamine-1-phosphate uridyltransferase, modulates its acetyltransferase activity. J. Mol. Biol. 386, 451–464 (2009).
Villarino, A. et al. Proteomic identification of M. tuberculosis protein kinase substrates: PknB recruits GarA, a FHA domain-containing protein, through activation loop-mediated interactions. J. Mol. Biol. 350, 953–963 (2005).
Dasgupta, A., Datta, P., Kundu, M. & Basu, J. The serine/threonine kinase PknB of Mycobacterium tuberculosis phosphorylates PBPA, a penicillin-binding protein required for cell division. Microbiology 152, 493–504 (2006).
Sajid, A. et al. Interaction of Mycobacterium tuberculosis elongation factor Tu with GTP is regulated by phosphorylation. J. Bacteriol. 193, 5347–5358 (2011).
Boitel, B. et al. PknB kinase activity is regulated by phosphorylation in two Thr residues and dephosphorylation by PstP, the cognate phospho-Ser/Thr phosphatase, in Mycobacterium tuberculosis. Mol. Microbiol. 49, 1493–1508 (2003).
Wheeler, P. R. et al. Functional demonstration of reverse transsulfuration in the Mycobacterium tuberculosis complex reveals that methionine is the preferred sulfur source for pathogenic Mycobacteria. J. Biol. Chem. 280, 8069–8078 (2005).
Parker, W. B. & Long, M. C. Purine metabolism in Mycobacterium tuberculosis as a target for drug development. Curr. Pharm. Des. 13, 599–608 (2007).
Jakubowski, H. The determination of homocysteine-thiolactone in biological samples. Anal. Biochem. 308, 112–119 (2002).
Corrales, R. M., Leiba, J., Cohen-Gonsaud, M., Molle, V. & Kremer, L. Mycobacterium tuberculosis S-adenosyl-l-homocysteine hydrolase is negatively regulated by Ser/Thr phosphorylation. Biochem. Biophys. Res. Commun. 430, 858–864 (2013).
Tuite, N. L., Fraser, K. R. & O'byrne, C. P. Homocysteine toxicity in Escherichia coli is caused by a perturbation of branched-chain amino acid biosynthesis. J. Bacteriol. 187, 4362–4371 (2005).
Roe, A. J., O'Byrne, C., McLaggan, D. & Booth, I. R. Inhibition of Escherichia coli growth by acetic acid: a problem with methionine biosynthesis and homocysteine toxicity. Microbiology 148, 2215–2222 (2002).
Arora, G. et al. Unveiling the novel dual specificity protein kinases in Bacillus anthracis: identification of the first prokaryotic dual specificity tyrosine phosphorylation-regulated kinase (DYRK)-like kinase. J. Biol. Chem. 287, 26749–26763 (2012).
Wang, F. et al. Mycobacterium tuberculosis dihydrofolate reductase is not a target relevant to the antitubercular activity of isoniazid. Antimicrob. Agents Chemother. 54, 3776–3782 (2010).
Sharma, K. et al. Transcriptional control of the mycobacterial embCAB operon by PknH through a regulatory protein, EmbR, in vivo. J. Bacteriol. 188, 2936–2944 (2006).
Bashiri, G., Rehan, A. M., Greenwood, D. R., Dickson, J. M. & Baker, E. N. Metabolic engineering of cofactor F420 production in Mycobacterium smegmatis. PLoS One. 5, e15803 (2010).
Khan, S. et al. Phosphorylation of enoyl-acyl carrier protein reductase InhA impacts mycobacterial growth and survival. J. Biol. Chem. 285, 37860–37871 (2010).
Gupta, M., Sajid, A., Arora, G., Tandon, V. & Singh, Y. Forkhead-associated domain-containing protein Rv0019c and polyketide-associated protein PapA5, from substrates of serine/threonine protein kinase PknB to interacting proteins of Mycobacterium tuberculosis. J. Biol. Chem. 284, 34723–34734 (2009).
Gomi, T., Takata, Y. & Fujioka, M. Rat liver S-adenosylhomocysteinase. Spectrophotometric study of coenzyme binding. Biochim. Biophys. Acta 994, 172–179 (1989).
Hershfield, M. S. Apparent suicide inactivation of human lymphoblast S-adenosylhomocysteine hydrolase by 2′-deoxyadenosine and adenine arabinoside. A basis for direct toxic effects of analogs of adenosine. J. Biol. Chem. 254, 22–25 (1979).
Jacobsen, D. W. et al. Rapid HPLC determination of total homocysteine and other thiols in serum and plasma: sex differences and correlation with cobalamin and folate concentrations in healthy subjects. Clin. Chem. 40, 873–881 (1994).
Kumar, A. et al. Homocysteine- and cysteine-mediated growth defect is not associated with induction of oxidative stress response genes in yeast. Biochem. J. 396, 61–69 (2006).
Arora, G. et al. Understanding the role of PknJ in Mycobacterium tuberculosis: biochemical characterization and identification of novel substrate pyruvate kinase A. PLoS One 5, e10772 (2010).
Sajid, A. et al. Phosphorylation of Mycobacterium tuberculosis Ser/Thr phosphatase by PknA and PknB. PLoS One 6, e17871 (2011).
Boyle, W. J., van der, G. P. & Hunter, T. Phosphopeptide mapping and phosphoamino acid analysis by two-dimensional separation on thin-layer cellulose plates. Methods Enzymol. 201, 110–149 (1991).
Larkin, M. A. et al. Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 (2007).
Pettersen, E. F. et al. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Acknowledgements
This work was supported by Council of Scientific and Industrial Research (CSIR)- funded project BSC-0104. We acknowledge Dr. Ghader Bashiri, University of Auckland, New Zealand for kindly providing the vector pYUBDuet. We thank Zachary Waldon (Proteomics Core Facility, Children's Hospital, Boston, MA) for identification of phosphorylation sites by mass spectrometry. We thank Dr. Sheetal Gandotra for her valuable suggestions and Dr. V. C. Kalia for critical reading of the manuscript.
Author information
Authors and Affiliations
Contributions
A. Singhal and G. A. conceived and designed the experiments. A. Singhal, G. A., A. Sajid, A. M., A. B., R. V. and S. U. performed the experiments. A. Singhal, G. A. and A. Sajid wrote the paper and prepared the figures. S. S., V. K. N. and Y. S. contributed reagents/materials/analysis tools. All authors read and approved the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Singhal, A., Arora, G., Sajid, A. et al. Regulation of homocysteine metabolism by Mycobacterium tuberculosis S-adenosylhomocysteine hydrolase. Sci Rep 3, 2264 (2013). https://doi.org/10.1038/srep02264
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep02264
This article is cited by
-
Structure, function and substrate preferences of archaeal S-adenosyl-l-homocysteine hydrolases
Communications Biology (2024)
-
Antioxidant molecular mechanism of adenosyl homocysteinase from cyanobacteria and its wound healing process in fibroblast cells
Molecular Biology Reports (2020)
-
Effect of Homocysteine on Biofilm Formation by Mycobacteria
Indian Journal of Microbiology (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
