
Abstract
Chiral nanoscale photonic systems typically follow either tetrahedral or helical geometries that require four or more different constituent nanoparticles. Smaller number of particles and different chiral geometries taking advantage of the self-organization capabilities of nanomaterials will advance understanding of chiral plasmonic effects, facilitate development of their theory and stimulate practical applications of chiroplasmonics. Here we show that gold nanorods self-assemble into side-by-side orientated pairs and “ladders” in which chiral properties originate from the small dihedral angle between them. Spontaneous twisting of one nanorod versus the other one breaks the centrosymmetric nature of the parallel assemblies. Two possible enantiomeric conformations with positive and negative dihedral angles were obtained with different assembly triggers. The chiral nature of the angled nanorod pairs was confirmed by 4π full space simulations and the first example of single-particle CD spectroscopy. Self-assembled nanorod pairs and “ladders” enable the development of chiral metamaterials, (bio)sensors and new catalytic processes.
Similar content being viewed by others


Introduction
Assemblies of gold nanoparticles (NPs)1,2,3,4 and nanorods (NRs)5,6 attract scientists by the diversity of optical plasmonic effects. Research in this area is concentrated around spectroscopy of different plasmon modes7, appearance of hot spots, enhancement of different optical effects by plasmonic fields8, plasmon-to-heat conversion and a variety of applications related to these effects9. Compared to the large progress in these areas, we are only beginning to understand a variety of chiral effects related to nanoscale plasmonic materials10,11,12,13,14,15,16,17. Chirality of individual NPs can arise from electronic “imprints” of molecules on their surface16,17. Different chirooptical effects for plasmonic materials could be unusually strong7,8,11,14,22 and enable several new photonics technologies11,7,23. The fundamental studies and technology of chiral photonics depends on finding new preparative methods for chiral plasmonic enantiomers. So far, it was primarily achieved using chiral stabilizers17 and chiral scaffolds forcing asymmetry in the geometrical positioning of NPs, while in other cases equal amounts of dextrorotatory (+) or levorotatory (−) enantiomers were formed20. NP/NR superstructures are typically designed to be tetrahedral10,21 or helical3,18,19,20 in geometry, which de facto replicates the cases of well-known chiral organic and biological molecules. Such geometries typically require more than four constituent NPs to construct chiral three-dimensional systems as well as complex biomolecules and multistep synthesis process. It would certainly be important to simplify methods of their preparation and to find different chiral geometries with smaller number of constituents.
In this paper, we demonstrate that strongly chiral nanoscale systems can be made from as few as two gold NRs. They form side-by-side oriented NR pairs as well as multi-NR “ladders”. Chiral properties originate from the small dihedral angle between two adjacent NRs which breaks centrosymetric nature of two parallel identical cylinders. Importantly, the synthesis proceeds via self-assembly route, which is characteristic for many nanoscale colloids and represents their intrinsic similarity to biomacromolecules24 and can be accomplished with different assembly triggers leading to both (+) and (−) enantiomers. The chiroplasmonic properties of NR pairs in dispersions match the CD spectra obtained for single NR pairs.
Results
Gold NRs were prepared by a standard seeded growth method25. Their assembly was triggered by the hybridization of DNA oligomers in the standard PCR process (Fig. 1A, see SI), as well as by simple addition of sodium citrate (SCI) or sodium carbonate (SCA). DNA “connectors” are chiral, while SCI and SCA assembly triggers are racemic and non-chiral, respectively.

Structural Characteristics of Nanorod Assemblies.
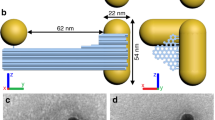
(A) Schematics of gold NRs dimers made with SCA, SCI and DNA. (B) Yields of NR dimers for SCA (black), SCI (red) and DNA (blue) assemblies, respectively, under optimized conditions and reaction times. (C–E) TEM images for dimers made by SCA (C), SCI (D) and DNA (E), respectively. (F–H) SEM images for NR dimers made by SCA (F), SCI (G) and DNA (H), respectively. (I–K) 3D TEM tomography images for front view (I), back view (J) and tilted view (K) of NR dimer assembled with SCA. (L,M) Schematics of (−) enantiomer (L) and (+) enantiomers (M). Geometrical parameters θ, d, s, denote twist angle between NRs (in deg), intra-nanorod gap (in nm) and vertical offset of NR with respect to each other (in nm), respectively. In most cases in this study, s was assumed to be negligible based on extensive evaluation of TEM images.
For all triggers, NRs assembled into dimers and larger “ladders” following the side-by-side (SbS) motif. NR dimers represent the basic element of such assemblies and determine their chirooptical effects. The dimers can be isolated both chemically and spectroscopically by judiciously selecting the aspect ratio (AR) of NRs and assembly conditions/time. The data presented below for DNA-connected dimers were obtained for NRs with length/diameter of 63.6 nm/22.0 nm, AR = 2.9. The data for SCI were obtained for NRs with length/diameter of 45.7 nm/21.8 nm, AR = 2.1, while SCA assemblies were investigated for NRs with length/diameter of 52.6 nm/22.0 nm, AR = 2.4. Chirooptical properties were characterized under conditions marked in Fig. 2 C, F, I unless otherwise noted. Statistical analysis of TEM/SEM images revealed that the yield of dimers at these conditions was as high as 74.5%, 78.2% and 71.1% for DNA, SCI and SCA triggers, respectively (Fig. 1B). When needed, we could also obtain other SbS assemblies, such as trimers, tetramers, pentamers, etc., as dominant products. The dimers, however, are the simplest and the most fundamental geometrical arrangement of NRs. Importantly, NR dimers may or may not have individual chirality26 and enantiomeric preference. Understanding of chirooptical properties of the dimers is quintessential for understanding of the same for other SbS assemblies27. Therefore, in this study, we shall focus on the experimental observations and theoretical understanding of their chirooptical behavior.

Optical Properties of Nanorod Assemblies.
UV-vis (A, D, G) and CD spectra (B, E, H) of NR assemblies and their temporal/concentration profiles (C, F, I) for specified assembly triggers and characteristic wavelengths. The time/concentration points for each spectrum are given and color-coded in each graph. λL, λT λp and λn, (blue lines and blue axes in C, F, I) denote spectral positions (in nm) of longitudinal (λL) and transverse (λT) peaks of NRs in the UV-vis spectra as well as spectral positions of positive (λp) and negative (λn) bands in CD spectra, respectively. Cp, Cnand similar notations (red lines and red axes in C, F, I) denote intensities (in mdeg) of main positive and negative plasmonic peak as well as peaks at specified wavelengths in CD spectra (C228, C219 and C290), respectively. Insets in A, D, G show the representative TEM images of NRs used for each assembly trigger. Time/concentration conditions characteristic for dominant presence of NR dimers are marked with green boxes in C, F, I.
TEM tomography allowed us to establish the detailed 3D geometry of NR dimers (Fig. 1 I–K). The two constitutive NRs are not parallel to each other, but have a distinct dihedral angle, θ (Fig. 1L, M). Aggregation of NR dimers into larger assemblies was greatly slowed down or prevented by their positive charge displaying electrokinetic potentials, ξ, of 15.3 mV, 20.2 mV and 14.1 mV for DNA, SCI and SCA NR dimers, respectively. The angled conformations of NR pairs dominate over the parallel one because conformations with θ ≠ 0 (Fig. 1) reduce their electrostatic repulsion and, therefore, are more thermodynamically favorable. Similar angled geometries due to balancing repulsive and attractive forces were also observed for many biopolymers and their nanoscale assemblies28.
The surface-to-surface gap between the NRs in the dimer, d (Fig. 1L), in solution was calculated from the dynamic light scattering (DLS) data describing the hydrodynamic diameter (Dh) along the short axis of the NR pair. For DNA, SCI and SCA-assembled dimers (Fig. S8 & S9), the corresponding d was 17.2 nm, 9.6 nm and 9.1 nm, respectively. In case of DNA, d is controlled by 50 base-pair DNA and is naturally larger than the inter-nanorod gap for NR dimers assembled with SCI and SCA. TEM tomography gave quite similar results although systematically smaller values for d, namely, 13.9 nm (DNA), 7.1 nm (SCI) and 6.3 nm (SCA) (Fig. S10) due to high-vacuum TEM conditions.
UV-vis spectra obtained for different reaction times (Fig. 2A, D) and trigger concentrations (Fig. 2G) show that the absorption peaks of the longitudinal bands for all three types of NRs undergo a progressively stronger blue shift by 8 nm, 11 nm and 7 nm for DNA, SCI and SCA, respectively, upon formation of the angled dimers. The transverse band in the UV-vis spectra shifts concomitantly to the red by 8 nm, 5 nm and 1 nm for the same triggers and same conditions. This observation correlates very well with their expected spectral behavior based on previous studies6,14.
The geometry of NR angled pairs makes them intrinsically chiral (Fig. 1 L, M) and natural dispersion of NRs in lengths further increases their asymmetry. The inequality of paired NR per se cannot, however, bring about chirality if θ = 0 and diminish the polarization rotation if θ ≠ 0 (Fig. S14). Simple geometrical considerations as well as electrodynamic calculations demonstrated that the rotation of plane-polarized light by the angled configuration of the NRs is indeed strongly chiral (Fig. S11–S16). For a given NR length and AR, the chiral optical activity is the highest for pairs from NR of equal length.
Chiral absorption bands appear in the plasmonic region of the UV-vis spectrum. The position and intensity of the bands is strongly dependent on the plasmonic coupling of dimer and the angle, θ, between the NRs (Fig. S18). These findings, however, do not necessarily mean that the chirality can be observed experimentally because of the expected thermodynamic equivalence of (+) and (−) enantiomers. Identical concentrations of the enantiomers make the optical media racemic and CD bands disappear.
The CD spectra of dimers revealed rich chiral photonic behavior (Fig. 2B, E, H) demonstrating that for all assemblies there is a strong preference for one specific enantiomer. DNA oligomers which are much shorter than origami-like templates11,3 have expected relatively weak CD peaks in the 200–250 nm region. Based on the previous studies, CD bands in the 500–800 nm region should be attributed to longitudinal and transverse plasmons of gold NRs9 with likely contribution of other (dark) modes. A strong correlation between the temporal trajectories of UV-vis absorption maxima of longitudinal (λL), transverse (λT) NR plasmons, growing intensities and spectral shifts of both positive (λp) and negative (λn) CD bands (Fig. 2C, F, I) can be clearly seen.
Unlike DNA, SCA and SCI have no chirooptical activity for the entire UV-vis window. Equally important, CD spectral signatures for NR dimers are virtually identical to those obtained for conditions with larger SbS assemblies (Fig. 2B, E, H). Therefore, the larger assemblies do not change the preference with respect to a specific enantiomer (Fig. S1–S3). Also note that the geometry of enantiomers must be different for the DNA- and SCI/SCA-triggered assemblies because the signs of the Cotton effect for the bisignated CD curves around the plasmonic bands in the 500–800 nm region are opposite (see SI). Specifically, the Cotton effect is positive for SCI and SCA NR assemblies and negative for NR pairs connected by DNA for the 620–800 nm window. Notably, the CD intensity for DNA connected NR dimers was stronger than dimers made by SCA and SCI in the 500–800 nm region. The intensive CD of NR dimers by DNA should originate from the stronger directionality of thermodynamic preference by ionic strength in DNA hybridization solution, which leads to larger excess of one enantiomer than the other.
The observed preference in (+)/(−) enantiomers for NR assemblies connected by DNA can potentially come from (a) an electronic “imprint” of the DNA helix on the plasmon oscillations in NRs, (b) some small amount of chiral impurity, (c) forced twisting of the angled NR dimers toward either (+) or (−) enantiomer by the DNA bridges and (d) intrinsic chirality of the produced NRs. Hypotheses (a) was rejected because we can see no correlation between CD bands corresponding to DNA strands at 200–250 nm and those at 610–640 nm and 740–790 nm related to the plasmon bands of NRs. The DNA bands change from positive to negative; they also shift initially to the blue and then to the red detailing dramatic change of DNA conformation in the course of NR assembly. If chirality of NR pairs is “borrowed” from DNA in the form of electronic imprint, these changes must be reflected in the plasmonic part of the spectrum (500–800 nm). However, this is not the case.
The observation of heterochiral NR assemblies induced by SCI and SCA confirmed this conclusion. CD spectra of SCI- and SCA-induced assemblies are similar to each other. Since SCA is achiral and SCI is racemic with no CD peaks on their own, the triggers cannot play the role of chiral “scaffolds” for these assemblies. The chiral selection of a specific enantiomer of SCI by cetyltrimethylammonium bromide (CTAB) coating to produce a particular enantiomer of the NR pair is difficult to envision. The new CD peaks, therefore, must reflect the enatiomeric preference of the NR themselves (Fig. 2 E, H).
We carefully considered hypothesis (b), i. e. the possibility of chiral impurities, as a potential reason for enantiomeric preference in NR pairs and, in particularly the possibility of the sergeant-and-soldier effect (SI). A model experiment with intentional addition of chiral admixtures, D- and L-threonine, showed that they did not result in any assembly nor affected the process and outcome of NR assembly with SCI (Fig. S4–S7).
Computer simulation of chiroptical properties can help in understanding the origin of chirality in the NR assemblies (Fig. 3). Expectedly, a parallel NR dimer of equal NRs showed negligible CD signals in the entire UV-vis window in the simulation because they are achiral. However, when θ ≠ 0, the simulations showed markedly different electromagnetic coupling efficiency with right and left circularly polarized light (RCP, LCP) (Fig. S17).

Experimental and Simulated Optical Properties of chiral NR dimers.
Experimental data (A, C, E) and corresponding simulation results (B, D, F) for NR dimers with θ = −9.5, 11, 14 deg, d = 17.1, 9.6, 9.1 nm and s = 0 for DNA-, SCI- and SCA-assemblies, respectively.
For simulations of angled NR pairs, the gap, d, between the rods was set to be 9.1 nm, 9.6 nm and 17.2 nm for SCA, SCI and DNA assemblies, respectively, according to the DLS data (see SI). Calculation of average θ from TEM and other experimental data are less reliable due to the sensitivity of θ to local/drying conditions. Therefore, we decided to consider θ as an adjustable parameter in the simulations. The angle θ was set to be negative and positive (Fig. 1 L, M) for DNA and SCI/SCA, respectively, according to their opposite signs of the Cotton effect (Fig. 2B, E, H). The offset, s was set to zero according to typical TEM images (Fig. 1 C–H) and extensive simulations which indicated its secondary role in determining chiroptical properties.
CD bands were calculated for NR dimers with various dihedral angles θ from +/−2 deg to +/−20 deg with a step of 1.5 deg (Fig. S18). The NR dimers with θ = −9.5 deg (DNA), 11 deg (SCI) and 14 deg (SCA) showed the best fit between simulated and experimental CD spectra in the 550–800 nm region (Fig. 3). The total ratio for (+) and (−) enantiomers, η (η(+), η(−)) can be calculated from molar CD spectrum combining experiments and simulations (see SI) to be ηDNA (20.9%, 79.1%), ηSCI (59.1%, 40.9%) and ηSCA (51.2%, 48.8%). In the simulation, the extinction spectrum of the dimer always showed a short wavelength peak (green line) and a long wavelength bump (blue line). The short and long wavelength peaks correspond to the antibounding and bounding modes of the dimer, respectively. The position of these peak also line up with the peaks of the short and long wavelength peaks of the CD spectra. The result is consistent with the single dimer we have measured (see below).
The simulated spectra for SCI- and SCA-triggered assemblies showed several weak bands at 215 nm, 300 nm and 510 nm (Fig. S19). They can be correlated with 219–228 nm, 290–300 nm and 500–520 nm bands observed experimentally (Fig. S20), although, as much stronger signals. These weaker bands are very similar for SCI- and SCA-assemblies in both position and sign and therefore, are not related to the trigger molecules but to dark plasmon modes of the SbS assemblies.
The strength and directionality of repulsive/attractive interactions in NRs determine the overall geometry of NR dimers. The result – the fine balance of repulsive-attractive interactions -- leads to thermodynamic preference of a specific enantiomer, which, we believe, originate from twisting of the angled NR dimers toward either (+) or (−) enantiomer in case of NR pairs connected by the chiral oligonucleotide bridges for DNA-triggered assemblies and weak but intrinsic chirality of organic coating of NRs that were made by using L-ascorbic acid in case of SCI and SCA assembly triggers (see Methods). The synthesized NRs showed a chiral band at 200–250 nm which should correspond to organic components of the NRs that can cause enantiomeric preference. The re-dispersed NR still have band at this region. This attribution of chiroplasmonic properties correlate well with the fact that DNA bridges result in (−) enantiomer while SCI/SCA give preference to (+) enantiomers. Some contribution in the chiral preference can also originate from non-ideal geometry of NRs that can deviate from the perfect cylinders as can be seen in TEM tomography images (Fig. 1I, J, K, see SI). In this case the enantiometric preference could originate from pairing of anisotropic NRs. Despite the difference in the type of preferred enantiomers, the chirality in all three systems and their photonic properties result from the twisting of one NR with respect to the other one which unifies all of them.
The optical properties of a single chiral dimer and the mechanism of chiral optical activity in our system were verified by single-particle spectroscopy. The first example of a dark-field scattering spectrum and CD spectrum for a single chiral dimer was acquired through isolating only one dimer in the area on a TEM grid (Fig. 4). The single dimer CD spectrum agreed very well with the ensemble CD spectrum in Fig. 3C. The positions of λp and λn were slightly blue shifted as it was likely that a larger NR dimer was selected arbitrarily to give a better signal.

Optical Properties of a Single NR Dimer.
(A) TEM images at different magnifications of a single twisted dimer and (B) correlated dark-field scattering image of the same single dimer as in (A). (C, D) Optical properties of a single SCI-assembled NR dimer as characterized by (C) the dark-field scattering spectrum and (D) the circular dichroism spectrum.
Discussion
SbS NR assemblies represent a new class of stereometamaterials and open the road to the realization of symmetry broken chiral photonics devices29 and new techniques of optical manipulation at nanoscale30. The first example of single particle CD measurements on NR pairs represents a particularly significant step in this direction which will also facilitate detailed understanding of chiroplasmonic effects. The demonstration of multiple enantiomers with controllable chiral preferences are essential for new routes for realization of metamaterials23, biosensing and chiral catalysis.
Methods
DNA-based NR assemblies were made using standard PCR protocol (see SI) starting from gold NRs surface-modified with appropriate PCR primers (see below) exclusively on their sides. The side-modification was carried out according to our previous work and Ref 14. In brief, 5 mL of as-made NRs (see SI) with AR = 2.9 and starting concentration of 0.24 nM dispersed in 0.005 M CTAB were centrifuged at 10000 RPM for 10 min. The supernatant was discarded while the precipitate was dissolved in 1 mL of 0.005 M CTAB. The end-sites of NRs were blocked by dithiothreitol (DTT) and thiol polyethylene glycol (PEG). DTT was added to NRs at a DTT/NR molar ratio of 10:1 and left to react for 8 h. NRs were centrifuged (7000 r/min, 10 min) and dissolved in 1 mL of 0.005 M CTAB. Then, PEG5000 was reacted with NRs at a PEG/NR molar ratio of 100:1, centrifuged (7000 r/min, 10 min) and dissolved in 1 mL of 0.005 M CTAB. The side-modification of NRs by either forward (Fd) or reverse (Rd) PCR primers was carried out in 500 μL of 0.005 M CTAB for 10 h at a primer/NR molar ratio of 400:1. The sequences for Fd and Rd primer were 5′-SH-TGGCTGACCCTGATGAGTTCG-3′ and 5′-SH-GGGCCATGATTACGCCAGTT-3′, respectively. After centrifugation (7000 RPM, 10 min), the modified NRs were redispersed in 500 μL of 0.005 M CTAB. Formation of NR pairs and other SbS assemblies was obtained by hybridization of complementary DNA strands obtained in a standard PCR process (see SI) performed with modified NRs in 50 μL of media containing 2.5 μL PCR buffer (50 M KCl, 10 mM Tris–HCl pH 9.0 at 25°C, 0.1% TritonX-100, 1.5 mM MgCl2), 1 μL of 1 mM deoxyribonucleoside triphosphates (dNTPS), 0.5 μL of Lambda DNA solution (0.25 ng) and 2.5 units of Taq Plus polymerase.
SCI-based NRs assemblies were made using NRs with AR = 2.1. Following centrifugation (11000 RPM, 10 min) to reduce the concentration of stabilizers and surface charge preventing self-assembly process, the NRs were re-dispersed in 500μL ultrapure water to have a concentration of 0.26 nM. The assembly process was triggered by adding 10 μL of 1.09 mg/ml SCI into 500 μL of NR dispersion (see SI). Aged NRs were obtained from the original 0.26 nM NR dispersion by incubation at room temperature for 3 days.
SCA-based NRs assemblies were made from NRs with AR = 2.4. After the centrifugation and dilution processes identical to those described above for SCI-based assemblies, the concentration of the NRs was 0.24 nM. The assembly process was triggered by adding different amounts of 2.92 mg/ml solution of SCA as described in Figure 2 to 500 μL of NR dispersion in ultrapure water.
References
Sardar, R. M., Funston, A., Mulvaney, P. & Murray, R. W. Gold Nanoparticles: Past, Present and Future. Langmuir 25, 13840–13851 (2009).
Elghanian, R., Storhoff, J. J., Mucic, R. C., Letsinger, R. L. & Mirkin, C. A. Selective colorimetric detection of polynucleotides based on the distance-dependent optical properties of gold nanoparticles. Science 277, 1078–1081 (1997).
Shen, X. B. et al. Rolling Up Gold Nanoparticle-Dressed DNA Origami into Three-Dimensional Plasmonic Chiral Nanostructures. J. Am. Chem. Soc. 134, 146–149 (2011).
Alivisatos, A. P. et al. Organization of 'nanocrystal molecules' using DNA. Nature 382, 609–611 (1996).
Liu, K. et al. Step-Growth Polymerization of Inorganic Nanoparticles. Science 329, 197–200 (2010).
Wang, L. B. et al. Side-by-Side and End-to-End Gold NR Assemblies for Environmental Toxin Sensing. Angew. Chem. Int. Ed. 49, 1–5 (2010).
Halas, N. J., Lal, S., Chang, W. S., Link, S. & Nordlander, P. Plasmons in Strongly Coupled Metallic Nanostructures. Chem. Rev. 111, 3913–3961 (2011).
Lieberman, I., Shemer, G., Fried, T., Kosower, E. M. & Markovich, G. Plasmon-Resonance-Enhanced Absorption and Circular Dichroism. Angew. Chem. Int. Ed. 47, 4855–4857 (2008).
Dreaden, E. C., Mackey, M. A., Huang, X., Kang, B. & El-Sayed, M. A. Beating Cancer to Death in Multiple Ways Using Nanogold. Chem. Soc. Rev. 40, 3391–3404 (2011).
Mastroianni, A. J., Claridge, S. A. & Alivisatos, A. P. Pyramidal and chiral groupings of gold nanocrystals assembled using DNA scaffolds. J. Am. Chem. Soc. 131, 8455–8459 (2009).
Kuzyk, A. et al. DNA-based self-assembly of chiral plasmonic nanostructures with tailored optical response. Nature 483, 311–314 (2012).
Sharma, J. et al. Control of Self-Assembly of DNA Tubules through Integration of Gold Nanoparticles. Science 323, 112–116 (2009).
Nykypanchuk, D., Maye, M. M., Van der Lelie, D. & Gang, O. DNA-guided crystallization of colloidal nanoparticles. Nature 451, 549–552 (2008).
Guerrero-Martínez, A. et al. Intense Optical Activity from Three-Dimensional Chiral Ordering of Plasmonic Nanoantennas. Angew. Chem. Int. Ed. 50, 5499–5503 (2011).
Hentschel, M., Schäferling, M., Weiss, T., Liu, N. & Giessen, H. Three-Dimensional Chiral Plasmonic Oligomers. Nano Lett. 12, 2542–2547 (2012).
Gautier, C. & Bürgi, T. Chiral Gold Nanoparticles. Chem Phys Chem 10, 483–492 (2009).
Qi, H. & Hegmann, T. Postsynthesis Racemization and Place Exchange Reactions. Another Step to Unravel the Origin of Chirality for Chiral Ligand-Capped Gold Nanoparticles. J. Am. Chem. Soc. 130, 14201–14206 (2008).
Wang, R. Y. et al. Chiral assembly of gold NRs with collective plasmonic circular dichroism response. Soft Matt. 7, 8370–8375 (2011).
Chen, C. L. & Rosi, N. L. Preparation of Unique 1-D Nanoparticle Superstructures and Tailoring their Structural Features. J. Am. Chem. Soc. 132, 6902–6903 (2010).
Srivastava, S. et al. Light-controlled self-assembly of semiconductor nanoparticles into twisted ribbons. Science 327, 1355–1359 (2010).
Chen, W. et al. Nanoparticle Superstructures Made by Polymerase Chain Reaction: Collective Interactions of Nanoparticles and a New Principle for Chiral Materials. Nano Lett. 9, 2153–2159 (2009).
Govorov, A. O., Fan, Z., Hernandez, P., Slocik, J. M. & Naik, R. R. Theory of Circular Dichroism of Nanomaterials Comprising Chiral Molecules and Nanocrystals: Plasmon Enhancement, Dipole Interactions and Dielectric Effects. Nano Lett. 10, 1374–1382 (2010).
Soukoulis, C. M. & Wegener, M. Past achievements and future challenges in the development of three-dimensional photonic metamaterials,. Nat Photonics. 5, 523 (2011).
Kotov, N. A. Inorganic Nanoparticles as Protein Mimics,. Science 330, 188–189 (2010).
Nikoobakht, B. & El-Sayed, M. A. Preparation and Growth Mechanism of Gold Nanorods Using Seed-Mediated Growth Method. Chem. Mater. 15, 1957 (2003).
Auguié, B., Alonso-Gómez, J. L., Guerrero-Martínez, A. S. & Liz-Marzán, L. M. Fingers Crossed: Optical Activity of a Chiral Dimer of Plasmonic Nanorods. J. Phys. Chem. Lett. 2, 846–851 (2011).
Husu, H. et al. Chiral coupling in gold nanodimers. Appl. Phys. Lett. 93, 183115 (2008).
Gibaud, T. et al. Reconfigurable self-assembly through chiral control of interfacial tension. Nature 481, 348–351 (2012).
Meng, X., Guler, U., Kildishev, A. V., Fujita, K., Tanaka, K. & Shalaev, V. M. Unidirectional Spaser in Symmetry-Broken Plasmonic Core-Shell Nanocavity. Scientific Reports 3, 1241 (2012).
Roxworthy, B. J. & Toussaint, K. C., Jr Plasmon nano-optical tweezers. Scientific Reports 2, 660 (2012).
Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (21071066, 91027038, 21101079, 21175034), the Key Programs from MOST (2012BAC01B07, 2012BAD29B05, 2012AA06A303, 2012BAD29B04, 2011BAK10B07, 2011BAK10B05, 2010AA06Z302, 2010DFB3047) and grants from Natural Science Foundation of Jiangsu Province, MOF and MOE (BE2011626, BK2010001, BK2010141). This material is based upon work partially supported by the Center for Solar and Thermal Energy Conversion, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number #DE-SC0000957. We acknowledge support from NSF under grant ECS-0601345; EFRI-BSBA 0938019; CBET 0933384; CBET 0932823; and CBET 1036672. The work is also partially supported by ARO MURI W911NF-12-1-0407 “Coherent Effects in Hybrid Nanostructures for Lineshape Engineering of Electromagnetic Media” (N.A.K. and S.L). The authors thank the University of Michigan's EMAL for its assistance with electron microscopy and for the NSF grant #DMR-9871177 for funding for the JEOL 2010F analytical electron microscope used in this work. Financial support from the Robert A. Welch Foundation (C-1664) is also acknowledged (S.L.). We would like to thank Sergio Dominguez-Medina and Jana Olson for help with the single particle measurements.
Author information
Authors and Affiliations
Contributions
C.X. and L.W. designed the experiments, interpreted and analyzed the data and co-wrote the paper. W.M., H.K., L.X., M.S. and Y.Z. performed the experiments, characterizations, prepared the samples and carried out computer calculations. Y.Z. and L.L. analyzed the data and carried out three-dimensional (3D) reconstruction of electron tomography. S.L., H.Z. and W.S.C. conceived and carried out single-particle CD spectroscopy experiments. N.A.K. conceived the project and experiments, analyzed the data, conceptualize the findings, figures, methods and co-wrote the paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Ma, W., Kuang, H., Wang, L. et al. Chiral plasmonics of self-assembled nanorod dimers. Sci Rep 3, 1934 (2013). https://doi.org/10.1038/srep01934
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep01934
This article is cited by
-
Photonically active bowtie nanoassemblies with chirality continuum
Nature (2023)
-
Self-assembled inorganic chiral superstructures
Nature Reviews Chemistry (2022)
-
Flexible synthesis of high-purity plasmonic assemblies
Nano Research (2021)
-
Chirality amplification by desymmetrization of chiral ligand-capped nanoparticles to nanorods quantified in soft condensed matter
Nature Communications (2018)
-
Analysis of the Optical Properties of Chiral Au Nanorod Stacks
Plasmonics (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
