
Abstract
An effective hemin catalyst on graphene support for selective oxidation of primary C-H bond in toluene is reported with an over 50% conversion rate achieved at mild conditions. Significantly this hybrid material shows catalytic efficiency in toluene oxidation with selectivity towards benzoic acid. The role of graphene support is discussed here as providing large contact area between the catalyst and the substrate, maintaining hemin in catalytically active monomer form, attracting electron to promote site isolation, as well as protecting hemin from oxidative degradation during the reaction. Moreover, graphene is suggested to largely alter the final product selectivity, due to the different π-π interaction strength between the graphene support and the substrate/oxidized products. With longer reaction time, overall conversion rate tends to maintain relatively unchanged while toluene undergoes a series of oxidation to convert mostly to benzoic acid.
Similar content being viewed by others


Introduction
Graphene is two-dimensional single layer material consisting of sp2-hybridized carbons1. It exhibits unique properties in various applications, such as energy storage and transfer devices, nanoelectronics and nanophotonic devices, nanocatalysts, etc.2,3,4,5. Particularly in catalytic research, using graphene oxide (GO) as starting material offers low cost route to synthesize graphene (GN) directly in solution form, hence greatly enhances its processability and capability of incorporation with other materials6,7,8,9,10,11,12. To date, there have been numerous reports demonstrating GN-Inorganic Nanomaterials based hybrid materials for different catalytic systems. For example, precious metal nanoparticles such as Au, Pd, Pt and Ag grown on GN are used as active catalyst for oxygen reduction reaction (ORR), oxidation reaction as well as hydrogenation reaction13,14,15,16. Ceramics nanomaterials like TiO2, MoS2 and Co3O4 on GN are found effective in organic pollutant photo-degradation, ORR and oxygen evolution reaction (OER) and H2 production in water splitting cell17,18,19,20,21. Still, to our best knowledge, GN-Organic Molecule based hybrid materials in catalytic application are not adequately explored except for a few reports22,23,24,25 and the influence of GN on organic catalyst is still unclear.
In contrast to inorganic catalysts, organometallic catalysts offer wider scope in reaction choices, better selectivity, lower cost and more flexible surface chemistry26,27. Among them metalloporphyrin has drawn substantial attention as an active catalyst to functionalize saturated C-H bonds with remarkable regioselectivity and enantioselectivity28,29,30,31. Hemin (iron protoporphyrin) is the active center of many heme-proteins32. Unlike inorganic catalysts, hemin suffers from instability upon oxidative degradation. It is reactive towards meso-cleavage via self-oxidation, thus transforms into catalytically inactive meso-hydroxyporphyrin derivatives30,33,34. Previous researchers put great efforts into structural modification in order to prevent this self-degradation29,30. Although these modifications indeed increase its activity and stability, it simultaneously increases synthetic difficulties and cost, which inevitably prevents its mass usage in industry. Alternatively, support materials are widely used in metalloporphyrin catalysts35,36,37,38,39. Nevertheless, those supports, like porous ceramics, polymers, nanofibers and nanotubes, are several magnitudes larger than metalloporphyrin molecule with limited active surface area. More importantly, metalloporphyrin connects to support mostly by covalent bond, which limits the choices of support materials and adds synthesis complexity29,39,40. Several recent papers demonstrate possibility of incorporating porphyrin with GN via π-π interaction22,23,24,25,41,42. To date, whether this hybrid material can maintain high activity, stability and selectivity in more challenging reactions, such as the saturated C-H bond oxidation reaction under higher temperature and longer reaction time to fulfill industry-oriented requirement is still unknown. More importantly, the function of GN in terms of overall catalyst performance and its influence over catalyst, such as the activity, selectivity and stability is still not well investigated.
Herein, we report hemin-graphene (H-GN) hybrid material synthesized via simple wet-chemical route as effective catalyst in saturated C-H bond oxidation reaction. Hemin is attached to GN via π-π interaction. The synthesis route is shown schematically in Figure 1 and details can be found in Supplementary S1. Toluene oxidation reaction is chosen to assess catalyst efficiency due to its commercial value and technological challenge. Toluene oxidation products benzyl alcohol (BAL), benzaldehyde (BAD) and benzoic acid (BAC) are important intermediates in pharmaceuticals, dyes, solvents, food preservative and so on. Current industrial method suffers from low conversion rate to maintain high selectivity43. Furthermore, addition of halogen cocatalysts and acidic solvents makes it environmentally unfriendly. Earlier studies44,45,46 have confirmed metalloporphyrin with or without support as active catalyst for this reaction. However, those results are either of relatively low yield, or from high temperature, high pressure condition, not mentioning the catalysts used are impractical to be synthesized in large quantity.

Schematic illustration of catalyst synthesis procedure and catalytic reaction involved.
(a), H-GN catalyst synthesis process and catalytic reaction. (b), Toluene oxidation reaction and main products catalyzed by H-GN.
Results
Materials characterization experiments: UV/Vis spectroscopy analysis, X-ray Photoelectron Spectroscopy (XPS) analysis, Atomic Force Microscopy (AFM) analysis and Raman spectroscopy analysis
GO and H-GN samples are characterized by UV-Vis spectroscopy (Fig. 2a). Yellowish brown colored GO dispersion displays an absorption peak at 231 nm with shoulder at 290–300 nm. For free hemin solution, the main adsorption peak is at 398 nm corresponding to Soret band and several small peaks between 500 nm and 700 nm from Q-bands23. After reduction, solution color changes to black and the broad peak at 265 nm shows the formation of GN47. Additionally, a second peak at 405 nm can be observed, indicating Soret band of hemin with bathochromic shift of 7 nm. This change can be reasoned as π-π interaction between the GN and the adsorbed hemin molecules, which is in agreement with previous reports that the π-π interaction between chemically converted graphene (CCG) and pyrene derivative leads to red shift of pyrene's characteristic adsorption peak48, as well as the observation of the red shift of the Soret band of the cationic porphyrin derivative upon its attachment to CCG44. The stability and thermal stability of H-GN is also tested (Supplementary Fig. S1). To conclude, hemin molecules are attached onto graphene via π-π interaction.

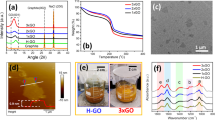
Materials characterization results using X-ray Photoelectron Spectroscopy (XPS), Atomic Force Microscopy (AFM) and Raman spectroscopy.
(a), UV-Vis spectra of GO, H-GN and hemin in ethanol. (b), XPS deconvolution of Fe2p in H-GN sample. UV-Vis spectra indicates after reduction, GO is reduced to GN and Hemin is attached onto GN via π-π interaction. XPS result indicates most Hemin molecules attached onto GN maintains monomer form. (c), AFM image of representative H-GN flakes, height profile with three selected line scan is indicated (blue, red and green lines). The average height is around 1.2 nm with average flake size of 3–5 μm. (d), The Raman shifts of GO, GN and H-GN.
XPS is another technique used to explore interaction between GN and hemin. GO sample survey shows no traceable signal of Fe2p at around 710 eV nor N1s at around 400 eV (Supplementary Fig. S2a). In contrast, the survey of H-GN shows the presence of both N1s and Fe2p signals, which is evidence of noncovalent functionalization of hemin on GN (Supplementary Fig. S2b). In C1s XPS spectrum of GO, two dominant peaks are at 284.5 eV and 286.4 eV, corresponding to C-C and C-O species, along with two weak peaks at 287.5 eV and 288.2 eV corresponding to C = O and O-C = O species (Supplementary Fig. S2c), respectively49,50. After reduction, the intensity of the peaks corresponding to C = O and O-C = O species reduces, due to the removal of the oxygen containing groups and the recovery of sp2 carbon network (Supplementary Fig. S2d). Meanwhile, a new peak at 285.7 eV rises due to the formation of C-N bonds from hydrazine hydrate51,52. In Fe2p XPS spectrum of H-GN (Fig. 2b), Fe 2p3/2 peak is much higher than Fe-O mixed peak (Fe-O bond), compared to that in H-GN sample prepared in water environment which typically results in dimerization of hemin (Supplementary Fig. S2e)53. Hence it confirms that most hemin molecules maintain monomer form on GN.
The morphology and thickness of H-GN is examined by AFM. From Supplementary Figure S3a, the thickness of the GN sheet is around 0.7 nm, which is consistent with previous reports of single-layer CCG7. The thickness of H-GN is determined as 1.2 nm with 0.5 nm increment compared with that of clean GN sheets (Fig. 2c). Considering hemin molecule height is around 0.2 nm54, it can be estimated hemin molecules are adsorbed onto GN sheet as monolayer or sub-monolayer on both sides without aggregation. Based on this monolayer attachment assumption, it can be calculated the coverage of hemin on GN is approximately 28.2%.
The interaction between GN and hemin molecules is also characterized by Raman spectroscopy (Fig. 2d). The initial GO structure shows two broad peaks at 1352 cm−1 and 1596 cm−1, corresponding to D and G bands. After conversion to GN, the D and G peaks become narrower and G peak shifts to 1600 cm−1. Moreover, D/G intensity ratio increases. This is due to increment of number of small sp2 domains and reduction of its average size7. More interestingly, upon attachment of hemin onto GN, both D and G peaks become broader compared to pure GN, with G-band shift to 1591 cm−1. It is clear that there is electron transfer between GN and hemin molecules42.
Catalytic activity results: overall catalytic efficiency test, reaction mechanistic study, optimization of reaction conversion and catalyst general applicability study
The catalytic efficiency of H-GN is measured in toluene oxidation reaction with a range of conditions. In view of vulnerability of hemin at high temperature and energy saving demand in industry, we adopt low temperature (60°C) and atmosphere pressure. The comparison of conversion process by different catalysts is shown in Figure 3a and summarized in detail in Table 1. Hemin by itself has peroxidase-like activity, however is unstable under oxidative environment33,34. Thus, its low activity (Table 1, Entry 2) in this reaction can be explained by quick degradation. In contrast, with protective support from GN, hemin seems to function well: H-GN yields appreciably higher conversion rate (9.42%) than hemin (1.02%) or GN (0.14%), or in absence of catalyst (0.22%) (Table 1, Entries 1–4). TON reaches 9,420 in 12 hours, which is comparable or even better than other reported catalysts (see Supplementary Table S5 for more comparison)44,45,46. In addition, it can be observed H-GN gives slightly different product profile from other systems that selectivity towards BAC is more significant than BAD and BAL. Similar phenomena are also observed when O2 is replaced by t-butyl hydroperoxide (TBHP) as oxidant (Supplementary Fig. S4 and Table S1). This indicates GN support somehow alters catalytic selectivity.

H-GN catalytic performance in Toluene oxidation reaction.
(a), Toluene conversion comparison with different catalysts using O2 as oxidant. The reaction temperature is 60°C and the reaction time is 12 hours. O2 is the oxidant and TBHP is the initiator. O2 pressure is 1 atm. Substrate/initiator molar ratio is 100. Substrate/catalyst molar ratio is 100,000. In GN sample, GN dosage is same as that in H-GN. (b), Toluene conversion rate and product selectivity using H-GN as catalyst. The reaction conditions are identical to (a) except reaction time is extended to 20 hours. (c), The toluene conversion rates of different catalyst dosages/additions. Reaction 1 (Rxn. 1) with one catalyst addition within 20 hours. Rxn 2 is with two additions within 20 hours. Rxn 3 is with five additions within 30 hours and Rxn. 4 is from seven additions within 30 hours. (d), Comparison of the final product selectivities among Rxn. 1 to Rxn. 4 shown in (c) and Rxn. 5 is the reaction with twelve additions within 48 hours. BAL = Benzyl Alcohol. BAD = Benzaldehyde. BAC = Benzoic Acid.
In order to monitor detailed conversion process, in-situ sampling is done during the reaction (Fig. 3a, b). The catalytic process can be separated into three stages35,45,46: firstly, with initial addition of H-GN catalyst, oxygen donors from peroxide oxidize hemin and forms oxoiron(IV) species. This induction period is slow and products are mainly BAL and BAD. Secondly, once oxoiron(IV) species accumulate to certain level and oxygen donors are continuously supplied from O2, activated Fe = O species start to catalyze oxidation efficiently. This active period is characterized by faster conversion process. At the end a saturation period was observed and is attributed to the deactivation of the hemin molecules through the reaction with the oxygen donors46 (black curve in Fig. 3a). Additionally, the increment of BAC is apparent (blue curve in Fig. 3b) while the percentage of BAD is decreasing (green curve in Fig. 3b).
To increase the conversion rate, we attempted to increase initial catalyst dosage (Supplementary Fig. S5). To our surprise, the addition of 5-fold catalyst at the beginning of catalytic experiment gives limited increment in conversion rate (12.94% vs. 10.27% in 20 hours) and longer induction period (4 hours vs. 3 hours). Further increasing catalyst dosage to 10-fold, instead of a higher conversion rate, a reduced conversion rate (9.15% in 20 hours) is observed. It might be attributed to the coagulation of concentrated GN sheets, which restricts the access of the substrates and oxidants to the hemin catalyst and increases the chances of hemin forming inactive μ-oxo dimer. To enhance the conversion, we tried adding same dosage of catalyst repeatedly (Fig. 3c). The second addition is at the 7th hour (1 hour prior to saturation period). Subsequent additions are done per 4 hours. Based on this method, considerable enhancement in conversion rate is achieved. After second addition, substrate conversion reaches 19.21% after 20 hours, compared to 10.27% from single addition reaction. In 30 hours, the reaction with five additions of H-GN results in 34.75% converted and seven times addition leads to a conversion rate of 40.56%. 48 hours reaction with twelve additions yields ultimate conversion rate of 50.14%, with TON of 4,170. More importantly, the selectivity towards BAC becomes more significant with increasing aliquots (94.28% of BAC after 48 hours with twelve times additions).
Furthermore, to demonstrate the general applicability of H-GN catalyst, ethylbenzene, cyclohexane and styrene are chosen as reference substrates (Supplementary Table S4). As ethylbenzene possesses lower C-H bond dissociation energy than toluene (357.3 kJ/mol vs. 368.2 kJ/mol)55, its conversion rate is higher than toluene (12.62% vs. 9.42%), as expected. Cyclohexane reaction gives 8.24% yield after 12 hours, with clear selectivity towards cyclohexanone. Similarly, H-GN results in 23.12% styrene conversion and 73.6% selectivity towards benzaldehyde. These results confirm that H-GN can serve as active catalyst for various oxidation reactions.
Discussion
Given these initial promising results, we investigated function of GN in H-GN system. As stated previously (Table 1 Entry 3, Fig. 3a), GN itself is inactive in toluene oxidation reaction. Hence we believe the main role of GN is to provide support for hemin. GN has remarkably high surface area and the distribution of hemin on its surface is even, thus sufficient contact between catalyst and substrate/oxidant can be expected. Secondly, it has been noted by previous report22 that the immobilization of hemin on GN can prevent hemin molecules to form catalytically inactive species by self-dimerization. Our XPS result on Fe2p spectrum of H-GN sample (Fig. 2b) further supports this statement. Thirdly, Raman Spectroscopy is used to study electron transfer between hemin and GN. From Raman spectrum of GN (Fig. 2d), it can be seen G band is at 1,601 cm−1. In contrast, G-band shifts to 1,592 cm−1 for H-GN. This is a result of electron transfer from hemin to GN42,56. GN serves as electron withdrawing species, which resembles halogen substitutes on meso-phenyl rings and β-positions of pyrroles in metalloporphyrins29. The halogens due to their electron-withdrawing nature have been reported to provide stronger site isolation of the central metal core, enhance electrophilicity of the metal-oxo entity, thus improve catalytic activity of metalloporphyrin29,30. Similar effect can be expected in hemin from the GN support. Lastly, GN prevents or greatly reduces the self-destruction process by blocking the attack from the oxygen donors to hemin's bare side, especially under oxidative conditions33,34.
Besides the protective effect, GN itself is also thought to influence the selectivity of products. With longer reaction time and larger amount of H-GN catalyst added, the production of BAC is more dominant and the production of BAL and BAD declined (Fig. 3d). One might argue that this change is due to thermodynamics, as stated in a previous research57. However, clearly different production trends are observed by evaluating the absolute productions of all three products in the H-GN catalyzed reactions, blank control reaction (without catalysts) and hemin catalyzed reaction.(Supplementary Tables S2, S3) In the blank reaction or the hemin catalyzed reaction (Supplementary Table S3a,b), the BAD and BAC productions always increase with reaction time, with the overall production of BAD always higher than that of BAC. Such a trend is totally different from the observations obtained in the H-GN catalyzed reactions (Supplementary Table S3c–f), where we can clearly see that the production of BAD increases in the beginning and then gradually declines toward the end of the reaction. It is also observed the overall production BAC increases and surpasses the production of BAD toward the end of the reactions. We consider this observed selectivity towards BAC in the H-GN catalyzed reaction as a result of the different interactions between the substituted aromatic compounds with the GN support. According to Rochefort58, the presence of electro-withdrawing substitute groups on benzene significantly modifies overall π-π interaction between adsorbed molecules and GN. The medium range attractive π-σ interaction overwhelms long-range repulsive π-π interaction, thus helps to bring electro-withdrawing groups closer to GN surface and causes stronger adsorption. In our case, BAC and BAD with -COOH and -CH = O has stronger tendency to be adsorbed onto GN surface, compared to BAL with –CH2OH group. And toluene is the least likely adsorbed. In the beginning of a reaction, active hemin molecules are abundant and toluene molecules are the majority, hence the primary adsorption of toluene on H-GN results in oxidation to BAL, which subsequently undergo oxidation to produce BAD and BAC. As a result, the yield of all three products increases with time. As the reaction proceeds, the amount of BAD rises, which is more competitive in preferential adsorption onto GN, thus leads to a higher chance for BAD to approach hemin and be oxidized to BAC. As-produced BAC may tend to haunt near GN surface, allowing only BAD with similar electrophilicity to adsorb, reducing the chances for less electrophilic species such as BAL and toluene to access the H-GN catalytic surface. This process produces BAC in expense of BAD, therefore promotes product profile change while maintaining a slower overall conversion rate increment. The phenomenon can be observed apparently from multiple addition reactions (see Fig. 4 for schematic illustration), especially after ∼22 hours, as BAC selectivity reaches nearly 100% after 12 additions of H-GN in 48 hours (Fig. 3d).

Schematic illustration of influence of GN on reaction process based on multiple addition reaction.
Interaction of substrate/products with GN support at the beginning of reaction process. Aromatic rings with more polar substitute groups (-COOH > -COH > -CHOH > -CH3) tend to have stronger adhesion to GN surface, subsequently block access of less polar counterparts. In the beginning (a), toluene molecule is majority, hence adsorption of toluene on H-GN to produce BAL is major reaction. Subsequently, oxidation to BAD (b) and BAC (c) is governing reaction. As a result, all three products dosages increase with time. With the reaction proceeds, the amount of BAD raises, which causes its preferential adsorption onto H-GN, thus a higher chance to approach hemin and form BAC (d). As-produced BAC will also haunt near GN surface, allowing only BAD with similar electrophilicity to adsorb, reducing chances for less electrophilic species such as BAL and toluene to access GN surface. This process produces BAC in expense of BAD, therefore promoting product profile change while maintaining overall conversion rate increment slower.
In conclusion, H-GN hybrid material is proved as highly effective catalyst in toluene oxidation reaction under mild conditions. GN serves as support to hemin catalyst by providing sufficient contact between substrate/oxidant and catalyst, maintaining hemin as catalytically active monomer form and preventing it from self-dimerization, promoting charge transfer from hemin to GN so as to enhance central iron core isolation and activity and protecting hemin from meso-cleavage via self-oxidation. GN itself also influences catalytic selectivity. Electro-withdrawing substitute groups on benzene promotes adsorption to GN, which results in higher chance for BAD with more electrophilic C = O group to approach hemin on GN and transform to BAC. Hence a significant selectivity towards BAC is observed especially with longer reaction time.
Methods
Catalyst preparation
Graphene oxide (GO) is prepared using modified Hummer's method. Aqueous GO solution is then transferred into ethanol before mixing with hemin solution in ethanol. Ammonia (28%) and hydrazine hydrate (35%) are then introduced. And the mixture is heated up at 65°C for 3.5 hours, following which, the H-GN in ethanol is washed with ethanol to remove hydrazine residue and unattached hemin molecules.
Catalytic reaction
Toluene is introduced into 25 ml round bottom flask in oil bath with condensation. While vigorously stirring, TBHP initiator is added and O2 gas (1 atm) is bubbled into solution continuously. After 1 hour, desired amount of catalyst is introduced into solution system and the solution is heated to 60°C. Sampling is done per 15 ± 1 min for the first hour and 30 ± 1 min afterwards.
Characterization techniques
UV/Vis analysis was completed on Beckman Coulter DU800. XPS tests were done with Kratos AXIS Ultra DLD spectrometer. AFM studies were performed using Bruker Dimension 5000 Scanning Probe Microscope. Raman analysis was done with Renishaw Microscope Raman Spectrometer. GC-MS analysis was done with Shimadzu GCMS QP2010Plus.
References
Novoselov, K. S. et al. Electric field effect in atomically thin carbon films. Science 306, 666–669 (2004).
Liu, Y. et al. Plasmon resonance enhanced multicolour photodetection by graphene. Nature Comm. 2, 579 (2011).
Yin, S. et al. Assembly of Graphene Sheets into Hierarchical Structures for High-Performance Energy Storage. ACS Nano 5, 3831–3838 (2011).
Lightcap, I. V., Kosel, T. H. & Kamat, P. V. Anchoring semiconductor and metal nanoparticles on a two-dimensional catalyst mat. storing and shuttling electrons with reduced graphene oxide. Nano Lett. 10, 577–583 (2010).
Li, Y. et al. Stabilization of high-performance oxygen reduction reaction Pt electrocatalyst supported on reduced graphene oxide/carbon black composite. J. Am. Chem. Soc. 134, 12326–12329 (2012).
Eda, G., Fanchini, G. & Chhowalla, M. Large-area ultrathin films of reduced graphene oxide as a transparent and flexible electronic material. Nat. Nanotechnol. 3, 270–274 (2008).
Tung, V. C., Allen, M. J., Yang, Y. & Kaner, R. B. High-throughput solution processing of large-scale graphene. Nat. Nanotechnol. 4, 25–29 (2009).
Huang, X., Qi, X., Boey, F. & Zhang, H. Graphene based composites. Chem. Soc. Rev. 41, 666–686 (2012).
Stankovich, S. et al. Graphene-based composite materials. Nature 442, 282–286 (2006).
Rao, C. N. R., Sood, A. K., Subrahmanyam, K. S. & Govindaraj, A. Graphene: the new two-dimensional nanomaterial. Angew. Chem. Int. Ed. 48, 7752–7777 (2009).
Huang, X. et al. Graphene-based materials: synthesis, characterization, properties and applications. Small. 7, 1876–1902 (2011).
Tan, C., Huang, X. & Zhang, H. Synthesis and applications of graphene-based noble metal nanostructures. Mater. Today. 16, 29–36 (2013).
Guo, S., Sun, S. FePt nanoparticles assembled on graphene as enhanced catalyst for oxygen reduction reaction. J. Am. Chem. Soc. 134, 2492–2495 (2012).
Hu, C. et al. Ternary Pd2/PtFe networks supported by 3D graphene for efficient and durable electrooxidation of formic acid. Chem. Comm. 48, 11865–11867 (2012).
Kou, R. et al. Stabilization of electrocatalytic metal nanoparticles at metal-metal oxide-graphene triple junction points. J. Am. Chem. Soc. 133, 2541–2547 (2011).
Liu, J., Fu, S., Yuan, B., Li, Y. & Deng, Z. Toward a universal "adhesive nanosheet" for the assembly of multiple nanoparticles based on a protein-induced reduction/decoration of graphene oxide. J. Am. Chem. Soc. 132, 7279–7281 (2010).
Liang, Y. et al. Co3O4 nanocrystals on graphene as a synergistic catalyst for oxygen reduction reaction. Nat. Mater. 10, 780–786 (2011).
Li, Y. et al. MoS2 nanoparticles grown on graphene: an advanced catalyst for the hydrogen evolution reaction. J. Am. Chem. Soc. 133, 7296–7299 (2011).
Du, J. et al. Hierarchically ordered macro-mesoporous TiO2-graphene composite films: improved mass transfer, reduced charge recombination and their enhanced photocatalytic activities. ACS Nano 5, 590–596 (2011).
Liu, J. et al. Self-assembling TiO2 nanorods on large graphene oxide sheets at a two-phase interface and their anti-recombination in photocatalytic applications. Adv. Func. Mater. 20, 4175–4181 (2010).
Xiang, Q., Yu, J. & Jaroniec, M. Synergetic effect of MoS2 and graphene as cocatalysts for enhanced photocatalytic H2 production activity of TiO2 nanoparticles. J. Am. Chem. Soc. 134, 6575–6578 (2012).
Xue, T. et al. Graphene-supported hemin as a highly active biomimetic oxidation catalyst. Angew. Chem Int. Ed. 51, 3822–3825 (2012).
Guo, Y. et al. Hemin-graphene hybrid nanosheets with intrinsic peroxidase-like activity for label-free colorimetric detection of single-nucleotide polymorphism. ACS Nano 5, 1282–1290 (2011).
Song, Y., Chen, Y., Feng, L., Ren, J. & Qu, X. Selective and quantitative cancer cell detection using target-directed functionalized graphene and its synergetic peroxidase-like activity. Chem. Commun. 47, 4436–4438 (2011).
Janan, M., Bao, Q., Yang, J. & Loh, K. P. Electrocatalytically active graphene porphyrin MOF composite for oxygen reduction reaction. J. Am. Chem. Soc. 134, 6707–6713 (2012).
D'Souza, D. M. & Muller, T. J. Multi-component syntheses of heterocycles by transition-metal catalysis. Chem. Soc. Rev. 36, 1095–1108 (2007).
Lee, J. et al. Metal-organic frameworks as catalysts. Chem. Soc. Rev. 38, 1450–1459 (2009).
Groves, J. T., Nemo, T. E. & Myers, R. S. Hydroxylation and epoxidation catalyzed by iron-porphine complexes. oxygen transfer from iodosylbenzene. J. Am. Chem. Soc. 101, 1032–1033 (1979).
Che, C., Lo, V. K., Zhou, C. & Huang, J. Selective functionalisation of saturated C–H bonds with metalloporphyrin catalysts. Chem. Soc. Rev. 40, 1950–1975 (2011).
Meunier, B. Metalloporphyrins as Versatile Catalysts for Oxidation Reactions and Oxidative DNA Cleavage. Chem. Rev. 92, 1411–1456 (1992).
Tabushi, T. Reductive dioxygen activation by use of artificial P-450 systems. Coord. Chem. Rev. 86, 1–42 (1988).
Zhang, G. & Dasgupta, P. K. Hematin as a peroxidase substitute in hydrogen peroxide determinations. Anal. Chem. 64, 517–522 (1992).
Chang, C. K. & Kuo, M. Reaction of iron(III) porphyrins and iodosoxylene. The active oxene complex of cytochrome P-450. J. Am. Chem. Soc. 101, 3413–3415 (1979).
Arasasingham, R. D., Balch, A. L., Cornman, C. R. & Latos-Grazynski, L. Dioxygen insertion into iron(III)-carbon bonds. NMR studies of the formation and reactivity of alkylperoxo complexes of iron(III) porphyrins. J. Am. Chem. Soc. 111, 4357–4363 (1989).
Evans, S. & Lindsay Smith, J. R. The oxidation of ethylbenzene by dioxygen catalysed by supported iron porphyrins derived from iron(III) tetrakis(pentafluoro- phenyl)porphyrin. J. Chem. Soc. Perkin Trans. 2, 174–180 (2001).
Balakrishnan, T. & Palani, T. Triphase catalysis: hydroxylation of cyclooctane by NaOCl catalyzed by coordinatively bound manganese(III) porphyrin to nitrogen based polymer supports. J. Appl. Polym. Sci. 77, 104–111 (2000).
Guo, C., Huang, C., Zhang, X. & Guo, D. Catalysis of chitosan-supported iron tetraphenylporphyrin for aerobic oxidation of cyclohexane in absence of reductants and solvents. Appl. Catal. A. 247, 261–267 (2003).
Rosa, I. L., Manso, C. M., Serra, O. A. & Iamamoto, Y. Biomimetical catalytic activity of iron(III) porphyrins encapsulated in the zeolite X. J. Mol. Catal. A: Chem. 160, 199–208 (2000).
Nakagaki, S. & Wypych, F. Nanofibrous and nanotubular supports for the immobilization of metalloporphyrins as oxidation catalysts. J. Colloid Interface Sci. 315, 142–157 (2007).
Fraile, J. M., Garcia, J. I. & Mayoral, J. A. Noncovalent immobilization of enantioselective catalysts. Chem. Rev. 109, 360–417 (2009).
Xu, Y. et al. Chemically converted graphene induced molecular flattening of 5,10,15,20-tetrakis(1-methyl-4-pyridinio) porphyrin and its application for optical detection of cadmium(II) ions. J. Am. Chem. Soc. 131, 13490–13497 (2009).
Geng, J. & Jung, H. Porphyrin functionalized graphene sheets in aqueous suspensions: from the preparation of graphene sheets to highly conductive graphene films. J. Phys. Chem. C. 114, 8227–8234 (2010).
Partnheimer, W. Methodology and scope of metal/bromide autoxidation of hydrocarbons. Catal. Today 23, 69–158 (1995).
Rebelo, S., Simoes, M., Neves, G. & Cavaleiro, J. J. Oxidation of alkylaromatics with hydrogen peroxide catalysed by manganese(III) porphyrins in the presence of ammonium acetate. Mol. Catal. A: Chem. 201, 9–22 (2003).
Huang, G. et al. An efficient oxidation of toluene over Co(II)TPP supported on chitosan using air. Catal. Lett. 114, 174–177 (2007).
Evans, S. & Lindsay Smith, R. The oxidation of ethylbenzene and other alkylaromatics by dioxygen catalysed by iron(III) tetrakis(pentafluorophenyl) porphyrin and related iron porphyrins. J. Chem. Soc. Perkin Trans. 2, 1541–1551 (2000).
Li, D., Muller, M. B., Gilje, S., Kaner, R. B. & Wallace, G. G. Processable aqueous dispersions of graphene nanosheets. Nat. Nanotechnol. 3, 101–105 (2008).
Xu, Y., Bai, H., Lu, G., Li, C. & Shi, G. Flexible graphene films via the filtration of water-soluble noncovalent functionalized graphene sheets. J. Am. Chem. Soc. 130, 5856–5857 (2008).
Paredes, J. I., Villar-Rodil, S., Martinez-Alonsso, A. & Tascon, J. M. D. Graphene Oxide Dispersions in Organic Solvents. Langmuir 24, 10560–10564 (2008).
Wu, Z. et al. Synthesis of high-quality graphene with a pre-determined number of layers. Carbon 47, 493–499 (2009).
Bai, H., Xu, Y., Zhao, L., Li, C. & Shi, G. Non-covalent functionalization of graphene sheets by sulfonated polyaniline . Chem. Commun. 1667–1669 (2009).
Su, Q. et al. Composites of graphene with large aromatic molecules. Adv. Mater. 21, 3191–3195 (2009).
Christ, B. V. Handbook of Monochromatic XPS Spectra, The Elements of Native Oxides (Wiley, New York, 2000).
Qutub, Y., Uzunova, V., Galkin, O. & Vekilov, P. G. Interactions of hemin with model erythrocyte membranes. J. Phys. Chem. B. 114, 4529–4535 (2010).
Gardner, K. A., Kuehner, L. L. & Mayer, J. M. Hydrogen atom abstraction by permanganate: oxidations of arylalkanes in organic solvents. Inorg. Chem. 36, 2069–2078 (1997).
Voggu, R., Das, B., Rout, C. S. & Rao, C. N. R. Effects of charge transfer interaction of graphene with electron donor and acceptor molecules examined using raman spectroscopy and cognate techniques. J. Phys.: Condens. Matter. 20, 472204–472208 (2008).
Kesavan, L. et al. Solvent-free oxidation of primary carbon-hydrogen bonds in toluene using Au-Pd alloy nanoparticles. Science 331, 195–199 (2011).
Rochefort, A. & Wuest, J. D. Interaction of substituted aromatic compounds with graphene. Langmuir 25, 210–215 (2009).
Acknowledgements
The authors acknowledge EICN, CNSI for AFM facilities and MIC for XPS and GCMS facilities. Y. H. acknowledges support from ARO proposal no. 54709-MS-PCS and NSF grant no. CBET-1033672.
Author information
Authors and Affiliations
Contributions
Y.H. and X.D. conceived the idea. Y.H. and Y.L. designed the experiments. Y.L. and X.H. performed the experiments. Y.L., Y.X., Y.W. and E.Z. participated in various aspects of the experiments and discussions. All authors contributed to the analysis of data and the writing of the manuscripts.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
supporting data and discussion
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Li, Y., Huang, X., Li, Y. et al. Graphene-hemin hybrid material as effective catalyst for selective oxidation of primary C-H bond in toluene. Sci Rep 3, 1787 (2013). https://doi.org/10.1038/srep01787
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep01787
This article is cited by
-
Solvent-free catalytic oxidation of toluene over heterogeneous CeMnOx composite oxides
Research on Chemical Intermediates (2022)
-
Oxidation of toluene to benzoic acid via VOTPP catalyst synthesized with an improved method
Monatshefte für Chemie - Chemical Monthly (2020)
-
Mimicking peroxidase active site microenvironment by functionalized graphene quantum dots
Nano Research (2020)
-
Interactions Between Hemin-Binding DNA Aptamers and Hemin–Graphene Nanosheets: Reduced Affinity but Unperturbed Catalytic Activity
Journal of Analysis and Testing (2019)
-
Synthesis of nitrogen- and iron-containing carbon dots, and their application to colorimetric and fluorometric determination of dopamine
Microchimica Acta (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
