
Abstract
Tissues are composed of diverse cell subpopulations each with distinct metabolic characteristics that influence overall behavior. Unfortunately, traditional histopathology imaging techniques are ‘blind’ to the spatially ordered metabolic dynamics within tissue. While mass spectrometry imaging enables spatial mapping of molecular composition, resulting images are only a static snapshot in time of molecules involved in highly dynamic processes; kinetic information of flux through metabolic pathways is lacking. To address this limitation, we developed kinetic mass spectrometry imaging (kMSI), a novel technique integrating soft desorption/ionization mass spectrometry with clinically accepted in vivo metabolic labeling of tissue with deuterium to generate images of kinetic information of biological processes. Applied to a tumor, kMSI revealed heterogeneous spatial distributions of newly synthesized versus pre-existing lipids, with altered lipid synthesis patterns distinguishing region-specific intratumor subpopulations. Images also enabled identification and correlation of metabolic activity of specific lipids found in tumor regions of varying grade.
Similar content being viewed by others


Introduction
Tissue is inherently heterogeneous, with overall metabolic function a result of a complex network of interactions between metabolically diverse cells and cell subpopulations. In many diseases, pathology is often accompanied by altered cellular metabolism in different regions throughout the affected tissue. In cancer, specifically, adaptive modifications in lipid metabolism are critical, with highly up-regulated de novo lipogenesis a characteristic feature of tumor cell metabolism1. As a class of molecules, lipids play a central role in cellular physiology and metabolism, with phospholipids being essential components of the cell membrane and required for cell proliferation2. Variations in lipid composition and degree of saturation/unsaturation of constituent fatty acid chains alter membrane fluidity and lipid-mediated cell functions, including activity of membrane-associated receptors, receptor binding, endocytosis and exocytosis3. Spatially-defined measurement of both lipid composition and turnover would provide critical information for probing the metabolic status of different cell subpopulations heterogeneously distributed throughout a tissue, which can then be linked to interrelated metabolic pathways (e.g. glycolysis, which provides pyruvate and acetyl-CoA precursors used in fatty acid synthesis)2. Quantifying lipid turnover, specifically which lipids are being newly synthesized versus pre-existing populations, provides information on active metabolic pathways and insight into how metabolism is adapting to sustain energy requirements4.
Unfortunately, traditional histopathology imaging techniques have limited capability to spatially resolve dynamic metabolic processes. Microscopy based on chemical stains and immunohistochemistry are optimal for assessing tissue morphology and detecting biomarkers, but these static measurements are ‘blind’ to the heterogeneity of metabolic dynamics. Therefore, these current imaging techniques need to be complemented by spatial measurements of molecular composition and metabolic flux.
Mass spectrometry imaging of tissue is a relatively new technique that has enabled mapping of the spatial composition of proteins, lipids and other metabolites5. Use of these techniques to spatially map lipids within tumors, specifically, has enabled the identification of lipid markers that correspond to tumor and non-tumor regions, lipid profiling of high-grade versus low-grade tumor regions, as well as the capability to classify tumor tissue by subtype and grade6,7,8. However, existing mass spectrometry methods capable of measuring metabolic flux typically utilize stable isotopic tracers, such as amino acids, glucose, etc. These are typically performed on homogenized tissue, resulting in a loss of spatial location and averaging of metabolite concentrations. More recently, integration of tracers with the mass spectrometry imaging modality time of flight-secondary ion mass spectrometry (TOF-SIMS) has been used that detects the spatial location of the stable isotopic tracer during tissue imaging (e.g. 15N thymidine)9. Although TOF-SIMS has the highest lateral resolution in mass spectrometry imaging, this technique relies on ion bombardment that results in molecular fragmentation – “elemental” localization (e.g. 15N) of isotopic tracers is possible, but the identity of the intact molecule into which the tracer was incorporated is lost. In contrast, a range of other mass spectrometry imaging techniques have been developed that currently have lower spatial resolution, but by utilizing ‘soft-ionization’ approaches this preserves molecular structure and makes them well-suited for imaging intact molecules5,10.
To address these limitations we developed kMSI, a technique for imaging metabolic turnover based on the in vivo incorporation of stable isotope-labeled precursors into intact, non-fragmented molecules. This approach couples soft desorption/ionization mass spectrometry imaging with in vivo metabolic labeling with deuterium to generate images of kinetic information of biological processes. In this work, kinetic information is considered to be the fraction of molecules isotopically incorporated with deuterium as a result of biosynthesis during a labeling period. Here we apply kMSI to a tumor, a tissue in which high growth rates and a diversity of metabolically transformed cells characterize the disease state. By isotopically labeling mice with deuterium and imaging excised tumors with nanostructure-initiator mass spectrometry (NIMS), we generated images of lipid kinetic information as a first step towards spatially resolving synthesis rates of intact molecules that distinctively characterize metabolically heterogeneous tumor cell subpopulations.
Results
In vivo biosynthetic incorporation of deuterium into tissue
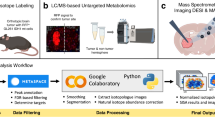
The kMSI workflow for generating and imaging deuterium labeled tissues is summarized in Figure 1. Briefly, administration of deuterated water (2H2O) to a tumor-bearing mouse provided known pathways for the biosynthetic incorporation of deuterium (2H) into tissue lipids ( Fig. 1A ). Via active metabolic pathways, newly synthesized lipids became isotopically labeled with deuterium, whereas pre-existing lipids remained unlabeled ( Fig. 2 ). By direct NIMS imaging of a deuterium-enriched tissue section, a unique mass spectrum was generated for each position for a variety of lipids ( Fig. 1B )10,11. Since each spectrum is a combination of isotopologues from unlabeled and 2H-labeled molecules at a given 2H2O enrichment, spectra could then be deconvoluted to identify and quantify specific unlabeled and newly synthesized lipids at each location throughout the tissue ( Fig. 1D ). This enabled determination of the relative amount of newly synthesized lipid during the period of label exposure as a fraction of total lipid, which could then be correlated with static histopathology-based findings, including spatially-distinct tissue morphology and cellular subpopulations ( Fig. 1C ).

Experimental workflow for using kMSI to define the spatial heterogeneity of lipid composition and biosynthesis.
(A) A tumor-bearing mouse is administered 2H2O-enriched water to incorporate deuterium into tissue as a result of active metabolism. (B) The deuterium-enriched tumor is excised, sectioned and imaged using NIMS. An individual mass spectrum is generated for each pixel every 50 μm, with spectra comprised of isotopologues from both 2H-labeled and unlabeled lipid molecules. (C) Serial sections of the tumor are used for histopathology correlation with kMSI results. (D) Deconvolution of spectra is performed to separate 2H-labeled and unlabeled lipids. Intensity images are generated to show the spatial distribution for both newly synthesized and pre-existing lipids.

Deuterium incorporation into phospholipids occurs during de novo fatty acid synthesis, a pathway utilizing substrates that have become metabolically deuterium-enriched themselves, including acetyl CoA, NADPH and water.
Hydrogen atoms highlighted in red indicate the location on a newly synthesized fatty acid where deuterium may replace hydrogen, as well as the locations on metabolic precursor molecules.
Isotopic incorporation of 2H into tissue was confirmed by comparing mass spectra generated from solvent-extracted tissue from a deuterium-treated versus an untreated mouse using liquid chromatography mass spectrometry (LC/MS) and NIMS. Isotopic patterns in spectra from the control, unlabeled mouse were consistent with naturally occurring isotopes; in contrast, isotopic patterns from the deuterium-treated mouse were composed of both 2H-labeled and unlabeled metabolites ( Supplementary Fig. S4 ). This is apparent by visual observation of the frequency in which the M1 isotopologue's intensity is greater than the M0 isotopologue's (monoisotopic mass) intensity12. As is typical in mass spectrometry, spectral patterns convey information on molecular composition; however, when tissue is also isotopically labeled, metabolic synthesis rates were also embedded within spectral patterns13.
Identification of phospholipids in mass spectrometry image data
By performing NIMS imaging directly on isotopically labeled and unlabeled thin tumor sections, individual mass spectra were generated for each location in 50 μm increments. Abundant ions (as measured by ion intensity) were detected between the mass range m/z 788-884, a typical mass range for phospholipids. Since no chromatography is performed in imaging experiments, multiple lipids of similar or identical masses may appear as a single ion [e.g. PC(34:01) with m/z = 760.5851 vs PE(P-38:00) with m/z = 760.6215]. Therefore to allow for multiple lipid species comprising what appears to be a single peak in the imaging data14 additional LC-MS and LC-MS/MS analysis was performed on solvent extracts of the tumor. Using exact mass, retention time and LC/MS/MS fragmentation spectra, these molecules were identified as being composed primarily of phosphatidylcholines (PCs), sphingomyelins (SMs), phosphatidylethanolamines (PEs) and phosphatidylserines (PSs). As is typical, phospholipids are detected as potassium adducts [M+K]+ in direct imaging of the tissue and as [M+H]+ in extracts15. Supplementary Table S2 provides a list of all lipids detected in this mass range, relative levels of each in the tumor extract and the difference (ppm) between the monoisotopic mass and detected peak m/z.
Validation of model of isotopic enrichment
Based on phospholipids, we developed a model of isotopic enrichment to apply spectral patterns to quantify both composition and new synthesis for each lipid. A full description of the model is provided in the Methods section. Briefly, the model relies on using an algorithm to fit the detected composite isotope pattern into the relative contributions arising from fully labeled and unlabeled lipids. To do this, the maximum number of deuterium atoms which are non-labile and capable of being biosynthetically incorporated on a lipid is represented by N. For the fully labeled lipid, these atoms are enriched in deuterium by the percent deuterium in body water. In this work, 4.5% of the body water was deuterium-enriched, resulting in a labeled lipid to be modeled as having N atoms enriched in deuterium by 4.5%. Fractional enrichment is given by the ratio of fully labeled lipid to total amount of that lipid species (labeled + unlabeled).
As shown in Figure 2, deuterium is incorporated into phospholipids during synthesis of fatty acid chains, with deuterium-enriched water contributing 2H instead of an H onto the growing fatty acid chain and modulating the isotopologue distribution pattern detected by mass spectrometry16. While the maximum theoretical number (N) of 2H's originating from water and capable of replacing H on a 16-carbon saturated fatty acid chain (palmitate) is N = 31, experimental values measured in vivo are typically lower depending on tissue type and other biological considerations17. Based upon previously reported values and experimental results, our model used N = 21 for palmitate synthesis, with N+/−3d (where d is the number of incremental 2-carbon units above or below a 16-carbon chain length) to account for elongation or unsaturation of fatty acid chains of each phospholipid18. Also, using cavity ringdown spectroscopy, body water was measured at 4.5 atom% 2H ( Supplementary Table S1 ) to allow calculation of a value for D, the fraction of exchangeable H's actually replaced by 2H during synthesis of a newly labeled molecule. These values for N ( Supplementary Table S2 ) and D were used to deconvolute the spectra and separate individual lipid species into two groups – unlabeled and 2H-labeled – reflecting pre-existing and newly synthesized lipids, respectively.
We validated our model and that selected parameters (Methods; Supplementary Fig. S3 ) were capable of discriminating between 2H-labeled versus unlabeled lipids. We further performed a sensitivity analysis to ensure that the relative abundance of one lipid species did not significantly influence the calculation of isotopic enrichment for other lipids (Supplementary Fig. S13). This resulted in the fraction of2H-labeled lipids in the control tumor to be quantified as near zero (within noise), while each lipid in the deuterium-treated tumor was found to be enriched in 2H-labeled isotopologues ( Supplementary Fig. S8, S9 ).
Heterogeneous distribution of lipid flux: newly synthesized vs. pre-existing lipids
To visualize the spatial distribution of newly synthesized versus pre-existing lipids, our model of isotopic enrichment was used to generate intensity images (2H-labeled and unlabeled) for each lipid species ( Supplementary Fig. S8, S9 ). Some of the most abundant lipids are shown for the deuterium-enriched tumor in Fig. 3. Interestingly, the spatial distribution of newly synthesized lipids did not correspond to that of pre-existing lipids. While some newly synthesized lipids localized in specific regions, others were distributed more heavily in the general periphery. This lipid-specific regional variation in flux demonstrates that active metabolic processes are region-specific and may correspond to cellular subpopulations or other characteristics of the tumor environment.

Intensity images for twelve selected phospholipids where each column represents a lipid and the three rows in each panel are top: unlabeled (pre-existing); middle: 2H-labeled (newly synthesized); and bottom: fraction labeled (2H-labeled/Total) (relative amounts of new versus pre-existing lipid levels).
The false color scale indicates the relative level of each lipid, maximally labeled, unlabeled or fraction labeled, distributed throughout the tissue. Image subscripts are provided to link to the comprehensive list of compounds characterized in this study (Supplementary Table S2, Supplementary Fig. 7,8). *Denotes a unique lipid with no shared mass. U = unlabeled, L = labeled, F = fraction labeled.
Identification of tumor sub-regions using kMSI spectra
To identify tumor sub-regions based upon both lipid composition and metabolic synthesis, K-means analysis was applied to each pixel of the tumor image. This analysis clustered pixels into 9 distinct patterns based upon spectral similarity19. Images and average spectra of pixels comprising each pattern tended to cluster spatially ( Fig. 4A, 4C ; Supplementary Fig. S5,S6 ). Based upon spectral patterns and spatial distribution, 3 regions were identified as artifact unrelated to tumor biology (IX - NIMS background ions; VII, VIII - embedding medium used in sectioning), while the remaining 6 were attributed to detection of endogenous tumor phospholipids. An overlay image shown in Fig. 4A shows the relative spatial distribution of each region. While Regions I-III localize in spatially-distinct areas (red, green and cyan), regions IV–VI overlap and appear more dispersed throughout the tumor periphery (yellow, blue and purple).

(A) Spatial distribution of clustered pixels for each K-means region attributed to the tumor phospholipids shown as individual images (left) and a merged overlay (right) with colors corresponding to the following regions: I-red, II-green, III-cyan, IV-yellow, V-blue, VI-purple.Scale bar, 5 mm. (B) H&E and Ki-67 stains corresponding to K-means regions I – III. Scale bars, 50 μm (H&E) and 100 μm (Ki-67). (C) Average spectra corresponding to K-means region I and II identified in the 2H-labeled tumor. Highlighted in gray are 3 areas that show different degrees of labeling for the same lipid between region I and II. This can be ascertained by visual inspection of the relative peak heights between adjacent peaks.
Correlation of lipid flux in tumor sub-regions with histopathology stains
To examine the potential biological basis for the K-means regions, a blinded histopathology examination was performed on an H&E serial section ( Supplementary Fig. S10–S12 ). Two of the tumor lobules were distinctly characterized by abnormal tissue architecture and nuclear pleomorphism associated with malignancy ( Fig. 4B , H&E I and II) which corresponded to K-means regions I and II. Further, these two regions could be morphologically distinguished from one another based upon observed degree of cell differentiation into glands, pleomorphic versus monomorphic character and range of cell size, with Region II showing more high-grade histologic features as compared to Region I.
Ki-67, a nuclear marker present in all stages of the cell cycle except G0, was also used to link kMSI results to cell proliferation20. Analysis indicated high rates of cell proliferation in both these regions, with region II higher than that of I ( Fig. 4B , Ki-67 I and II) and region II also having the highest fractional amount of newly synthesized lipids ( Supplementary Fig. S7c ). Necrotic areas of the tumor were also identified in the H&E stain, which corresponded with region III, the region with lowest levels of new synthesis ( Fig. 4B ; Supplementary Fig. S7b,c ). Although regions IV–VI were difficult to characterize in terms of tumorigenic properties in relation to the H&E stain, alterations in lipid composition were clearly reflected by the mass spectrometry imaging analysis. This suggests that Regions IV–VI may be composed of multiple cell subpopulations interspersed with each other and this kinetic histology approach could provide a mechanism for distinguishing each one.
Discussion
Further insight was gained by examining synthesis of individual lipids ( Fig. 3 ) with respect to each K-means region ( Fig. 4A ). Some of the most abundant phospholipids identified in the imaging data were those containing saturated and monounsaturated fatty acids (SFAs and MUFAs), including PC (34:0), PC (34:1), PC (36:1) and PS (36:1). Synthesis of these fatty acids is typically upregulated in aggressive tumors, with SFAs being the initial product of de novo lipogenesis and conversion to MUFAs occurring by only a single desaturation reaction21. The balance of SFAs to MUFAs has been implicated as critical for tumor cell survival, such that when this balance is shifted the cancer cell cannot survive22. Synthesis of phospholipids containing SFAs and MUFAs occurred heterogeneously within the tumor. While highest levels of PC (34:0) were associated with K-means Region I, PC (34:1) and PC (36:1) were distributed more throughout the tumor periphery in Regions IV–VI. Further, K-means Region II, the most high grade area identified within the tumor, had only minimal amounts of many of these lipids, but was highly abundant in PC (36:3), with three lipid species PC (38:3), PS (36:1) and PS (38:4) newly synthesized primarily in this region. These results suggest that region I and II, both high grade regions, may be composed of discrete cell subpopulations relying on alternate metabolic pathways for survival. Additionally, synthesis of phospholipids containing highly polyunsaturated fatty acids (PUFAs) were not generally localized in regions I and II, where the SFAs and MUFAs were found, implying that lipid desaturation was region specific. This may be due to differential expression of fatty acid synthase (FAS) and desaturase enzymes between cell populations or levels of incorporation of dietary fatty acids. Specifically, new synthesis of PC (38:6) and PC (38:7), containing 6 and 7 unsaturated bonds, respectively, were found throughout the tumor periphery in Regions IV–VI ( Fig. 3 ). PUFAs change membrane fluidity and structure, a compositional change that modifies overall cell function and response3. Further, PUFAs have been shown to be particularly toxic to certain cancer cell lines given their susceptibility to lipid peroxidation and increasing cytotoxic oxidative stress within the tumor cell23,24, suggesting that the spatial distribution of PUFAs within the tumor may correspond to additional cell subpopulations with diverse metabolic characteristics and level of aggressiveness.
These results show that kMSI is capable of generating kinetic images that distinguish metabolically distinct tissue regions. In this case, differences in de novo lipogenesis and phospholipid composition were found to correspond to histopathologically-distinct regions in the tumor. These observations are consistent with recent studies showing intratumor heterogeneity between cell subpopulations in terms of genetics and metabolism, with different therapeutic approaches necessary owing to these differences25,26,27,28,29,30. Since deuterium administration in the form of 2H2O is already commonly applied in the clinical setting14, kMSI images of the dynamic metabolic processes within tissues can complement existing histopathology techniques and may find broad utility in understanding physiology, disease and development.
Methods
Animal care, deuterium administration and tissue collection
Solid mammary tumors were obtained by transplanting Trp53-null mammary epithelium fragments (Balb/c background) into the cleared fat pad of F1 backcross female mice generated by the female interspecific F1 hybrid mice between BALB/c and SPRET/EiJ crossing with BALB/c male mice. 2H2O was administered by injecting a 30 mL/kg intraperitoneal bolus dose of sterile 99.9% 2H2O + 0.1% NaCl13, followed by free access to drinking water (8% 2H2O) and standard mouse chow. Animals were euthanized 5 days after 2H2O administration, then mammary tumor and serum collected and immediately flash-frozen on dry ice and stored at −80°C. As an unlabeled control, tumor and serum were also collected from a mouse never given 2H2O. Animal treatment and care was performed in accordance with animal protocols approved by the Animal Welfare and Research Committee at Lawrence Berkeley National Laboratory (AUP 9111 & 27010).
NIMS imaging
For each tumor, half was embedded in OCT medium and sectioned at −18°C using a Leica 1950S cryostat and the other half used for lipid extraction. Tumor cross-sections 5 μm thick were thaw-mounted onto a NIMS chip in preparation for MSI. Image acquisition was performed using a 5800 TOF/TOF (AbSciex, Foster City, CA) in positive reflector MS mode, with an Nd-YAG laser (200 Hz, 4650 laser intensity) acquiring spectra over a range of 500–1500 Da (900 Da focus mass) and accumulating 18 shots/spot. The 4800 Imaging Tool software was used to raster across the sample and record spectra in 50 μm × 50 μm step-sizes. Data viewing and image reconstruction was performed using custom software developed using the Matlab (Mathworks, Natick, MA) programming language.
Metabolite extraction
Frozen tumor samples (~4 mm × 4 mm) were dried in a FreeZone 2.5 lyophilizer (Labconco, Kansas City, MO) (20–50 mg dry weight), homogenized using a Mini-Beadbeater (BioSpec Products, Bartlesville, OK) for 5 seconds, then further homogenized in 350 μL MeOH:IPA:H2O (3:3:2 v/v) for 4 seconds (2x). Samples were centrifuged at 15,000 rpm for 2 minutes, supernatant collected, filtered and stored at −80°C.
NIMS of tumor extracts
Sample extracts dissolved in MeOH:IPA:H2O (3:3:2 v/v) were spotted directly onto a NIMS chip in 0.5 μL drops, air-dried 30 seconds, then excess sample wicked off. Mass spectra were obtained using a 5800 TOF/TOF (AbSciex, Foster City, CA) in positive reflector MS mode with an Nd-YAG laser (200 Hz, 2950 laser intensity). Spectra were acquired over a range of 250–1500 Da (900Da focus mass) and accumulating 18–22 shots/spot.
Liquid Chromatography Electrospray Ionization-Mass Spectrometry (LC ESI-MS) and MS/MS of tissue extracts
MS of tumor extracts was performed via LC-MS using both normal and reverse phase chromatography and MS and MS/MS data were collected using an Agilent ESI-QTOF. Normal phase used a 2.1 mm × 150 mm 1.7 μm Acquity UPLC BEH Amide HILIC column (Waters Corporation, Milford, MA). Here,chromatography was performed at a flow rate of 40 μL/min using a 2 or 4 μL sample injection volume. The column was equilibrated with 100% buffer B (90% acetonitrile w/5 mM ammonium acetate) for 5 minutes, diluting buffer B down to 45% with buffer A (H2O w/5 mM ammonium acetate) over 30 minutes and finally isocratic elution in 45% buffer B for 10 minutes. Reverse phase chromatography used a 5 μm 150 × 0.5 mm Zorbax SB-C18 column (Agilent) at a flow rate of 30 μL/min using a 1 μL injection volume. The column was equilibrated with 80% buffer A (60:40 H2O:ACN w/5 mM ammonium acetate) for 2 minutes, diluting buffer A down to 65% with buffer B (90:10 IPA:ACN w/5 mM ammonium acetate) over 8 minutes, down to 25% A over 22 minutes, followed by isocratic elution in 99% buffer B for 6 minutes. For LC/MS/MS, fragmentation data was acquired using 10, 20 and 40 V collision energies.
Histopathology stains
Staining was performed on 10 μm thick tumor serial sections taken within 300 μm of the NIMS-imaged section. Chemical stains included hematoxylin and eosin (H&E), which differentiated cell nuclei (blue) from cytoplasm and protein (red). An immunohistochemical HRP/DAB stain (brown) for the cell proliferation marker Ki67 was also performed using monoclonal [SP6] to Ki67 (abcam,Cambridge, MA; #ab16667). Digital images were acquired using a ScanScope XT (Aperio, Vista, CA). Stains were performed both in-house and by the UCSF Helen Diller Family Comprehensive Cancer Center, Immunohistochemistry Core Facility.
Deuterium enrichment in serum
Serum samples were analyzed for the amount of deuterium in body water by cavity ringdown spectroscopy using a Liquid Water Isotope Analyzer with an automated injection system, version 2 upgrade (Los Gatos Research, Mountain View, CA). Deuterium-enriched serum was diluted 1:100 prior to injection while unenriched serum was analyzed without dilution. Each sample was injected 6 times and the average of the last three measurements used for data analysis. A standard curve was run before and after samples for calculation of deuterium enrichment. Intra-run precision was less than 2 delta per mil (parts per thousand) and inter-run precision was less than 3.5 delta per mil. This analysis was performed by Metabolic Solutions, Inc. (Nashua, NH).
NIMS wafer fabrication
Preparation of a NIMS surface has been thoroughly described elsewhere30,31. Briefly, silicon wafers (Silicon Quest International, Santa Clara, CA) (single-sided polished P/Boron, orientation <1 0 0 >, resistivity 0.01–0.02 Ω cm, 525 +/− 25 μm thick) were electrochemically etched using 25% hydrofluoric acid in ethanol under a constant current of 2.4A for 15 minutes, then coated with bis(heptadecafluoro-1,1,2,2-tetrahydrodecyl) tetramethyl-disiloxane (Gelest, Morrisville, PA) as initiator.
Mass spectrometry imaging data processing
Raw spectra with m/z values in the range of 790–880 were imported from each image file (.img format). Representative peaks with high average intensity were correlated with the theoretical mass of the identified tumor lipids ( Supplementary Table S2 ). Exact masses of identified lipid species were then used to calibrate m/z values comprising the full spectra ( Supplementary Fig. S1 ). All further analyses were performed on these calibrated spectra.
Model of isotopic enrichment
Since the final measured spectrum is a composite of both2H-labeled and unlabeled isotopic patterns from the detected lipids ( Supplementary Fig. S2 ), each spectrum comprising a pixel was deconvoluted to separate each lipid species into two groups – unlabeled and 2H-labeled – reflecting pre-existing and newly synthesized lipids, respectively. An unlabeled lipid was modeled as having an isotopic pattern specified by chemical formula and naturally-occurring distribution of heavy isotopes ( Supplementary Fig. S2 – F1 ); the 2H-labeled lipid was modeled as having an isotopic pattern modulated by an increased amount of heavy hydrogen isotopes (2H replacing H) due to metabolic incorporation from 2H-enriched water ( Supplementary Fig. S2 – F2 ), with the modulation determined by (i) the amount of deuterium measured in enriched body water, atom% 2H (D) and (ii) N, the maximum number of deuterium atoms capable of being incorporated into newly synthesized lipids originating from water18.
Model isotopic patterns were generated using the fast Fourier transform-based method32,33 in which isotopic abundances for any given elemental isotopic composition are user-specified. A fitting algorithm, implemented in Matlab 2011b, finds a coefficient, xi, for each term, F1i and F2i, in the model (i.e.: 45 unlabeled and 45 2H-labeled patterns) that minimizes the difference between the model and measured spectra using non-negative least squares fitting: Minimize x in  , where x ≥ 0, F is the matrix of spectrum for the model patterns and M is a vector of the measured spectra at a given position34. For each pixel spectrum, this was used to calculate the relative fraction of newly synthesized species.
, where x ≥ 0, F is the matrix of spectrum for the model patterns and M is a vector of the measured spectra at a given position34. For each pixel spectrum, this was used to calculate the relative fraction of newly synthesized species.
In this model, values of N and D were based on experimental parameters as well as previously reported values from literature arising from biological considerations and experimental results16,18,35. Deuterium incorporation during synthesis of fatty acids is summarized in Figure 2, with elongation and desaturation reactions increasing or decreasing the value of N16. During de novo lipogenesis, hydrogen molecules are derived from metabolic precursors already enriched from 2H2O through various metabolic pathways and contribute 2H instead of an H onto the growing fatty acid chain in proportion to the 2H/H ratio in body water35. While theoretically the maximum number of 2H's on a C-16 saturated fatty acid chain (palmitate) is N = 31, experimental values measured in vivo are typically lower depending on tissue type and other biological considerations17. Also, since choline is primarily dietary-derived, hydrogen atoms on the phosphocholine headgroup contribute minimally to labeling36. Our model used D = 4.5atom% and N = 21 for palmitate synthesis, with N+/-3d (d is the number of incremental 2-C units above or below C-16) to account for elongation or unsaturation18. This model does not consider hydrogen atoms bound to heteroatoms that are labile and therefore in exchange with H in natural abundance during sample processing.
Kinetic images of lipid flux patterns
Kinetic images for individual lipids ( Fig. 4 ) were generated based on the relative contribution of the 2H-labeled vs. unlabeled population in the measured spectra for each pixel. Described earlier, our model of isotopic enrichment calculated the total summed intensity in the measured spectra resulting from all isotopologues arising from each individual lipid, in either the 2H-labeled or unlabeled form. In Figure 4, all intensity values for each lipid (shown by the color bar) are normalized to the same value.
K-means clustering
Pixels comprising the mass spectrometry image were grouped into 9 regions by K-means analysis using a correlation distance function in Matlab 2011b19. This identified and grouped pixels with similar spectral patterns based on having similar relative intensities for the detected ions. This process was repeated 10 times and the iteration with the smallest error used for analysis.
References
Swinnen, J. V., Brusselmans, K. & Verhoeven, G. Increased lipogenesis in cancer cells: new players, novel targets. Current opinion in clinical nutrition and metabolic care 9, 358–365 (2006).
Abramson, H. N. The lipogenesis pathway as a cancer target. Journal of medicinal chemistry 54, 5615–5638 (2011).
Spector, A. A. & Yorek, M. A. Membrane lipid composition and cellular function. Journal of lipid research 26, 1015–1035 (1985).
Deberardinis, R. J., Sayed, N., Ditsworth, D. & Thompson, C. B. Brick by brick: metabolism and tumor cell growth. Current opinion in genetics & development 18, 54–61 (2008).
Schwamborn, K. & Caprioli, R. M. Molecular imaging by mass spectrometry--looking beyond classical histology. Nat Rev Cancer 10, 639–646 (2010).
Wiseman, J. M., Puolitaival, S. M., Takats, Z., Cooks, R. G. & Caprioli, R. M. Mass spectrometric profiling of intact biological tissue by using desorption electrospray ionization. Angew Chem Int Ed Engl 44, 7094–7097 (2005).
Willems, S. M. et al. Imaging mass spectrometry of myxoid sarcomas identifies proteins and lipids specific to tumour type and grade and reveals biochemical intratumour heterogeneity. The Journal of pathology 222, 400–409 (2010).
Eberlin, L. S. et al. Classifying human brain tumors by lipid imaging with mass spectrometry. Cancer research 72, 645–654 (2012).
McMahon, G., Glassner, B. J. & Lechene, C. P. Quantitative imaging of cells with multi-isotope imaging mass spectrometry (MIMS)-Nanoautography with stable isotope tracers. Appl Surf Sci 252, 6895–6906 (2006).
Northen, T. R. et al. Clathrate nanostructures for mass spectrometry. Nature 449, 1033–1036 (2007).
Yanes, O. et al. Nanostructure initiator mass spectrometry: tissue imaging and direct biofluid analysis. Anal Chem 81, 2969–2975 (2009).
Diraison, F., Pachiaudi, C. & Beylot, M. In vivo measurement of plasma cholesterol and fatty acid synthesis with deuterated water: determination of the average number of deuterium atoms incorporated. Metabolism: clinical and experimental 45, 817–821 (1996).
Turner, S. M. et al. Measurement of TG synthesis and turnover in vivo by 2H2O incorporation into the glycerol moiety and application of MIDA. American journal of physiology. Endocrinology and metabolism 285, E790–803 (2003).
Murphy, R. C., Hankin, J. A. & Barkley, R. M. Imaging of lipid species by MALDI mass spectrometry. Journal of lipid research 50 Suppl S317–322 (2009).
Hankin, J. A. & Murphy, R. C. Relationship between MALDI IMS Intensity and Measured Quantity of Selected Phospholipids in Rat Brain Sections. Anal Chem 82, 8476–8484 (2010).
Wadke, M., Brunengraber, H., Lowenstein, J. M., Dolhun, J. J. & Arsenault, G. P. Fatty acid synthesis by liver perfused with deuterated and tritiated water. Biochemistry 12, 2619–2624 (1973).
Lee, W. N. P. et al. In-Vivo Measurement of Fatty-Acids and Cholesterol-Synthesis Using D2o and Mass Isotopomer Analysis. Am J Physiol 266, E699–E708 (1994).
Ajie, H. O. et al. In-Vivo Study of the Biosynthesis of Long-Chain Fatty-Acids Using Deuterated Water. Am J Physiol-Endoc M 269, E247–E252 (1995).
Jones, E. A. et al. Multiple statistical analysis techniques corroborate intratumor heterogeneity in imaging mass spectrometry datasets of myxofibrosarcoma. Plos One 6, e24913 (2011).
Dowsett, M. et al. Assessment of Ki67 in breast cancer: recommendations from the International Ki67 in Breast Cancer working group. Journal of the National Cancer Institute 103, 1656–1664 (2011).
Hilvo, M. et al. Novel theranostic opportunities offered by characterization of altered membrane lipid metabolism in breast cancer progression. Cancer research 71, 3236–3245 (2011).
Igal, R. A. Stearoyl-CoA desaturase-1: a novel key player in the mechanisms of cell proliferation, programmed cell death and transformation to cancer. Carcinogenesis 31, 1509–1515 (2010).
Maheo, K. et al. Differential sensitization of cancer cells to doxorubicin by DHA: a role for lipoperoxidation. Free radical biology & medicine 39, 742–751 (2005).
Bougnoux, P., Hajjaji, N., Maheo, K., Couet, C. & Chevalier, S. Fatty acids and breast cancer: sensitization to treatments and prevention of metastatic re-growth. Prog Lipid Res 49, 76–86 (2010).
Bertos, N. R. & Park, M. Breast cancer - one term, many entities? The Journal of clinical investigation 121, 3789–3796 (2011).
Marusyk, A. & Polyak, K. Tumor heterogeneity: Causes and consequences. Bba-Rev Cancer 1805, 105–117 (2010).
Tennant, D. A., Duran, R. V., Boulahbel, H. & Gottlieb, E. Metabolic transformation in cancer. Carcinogenesis 30, 1269–1280 (2009).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Gerlinger, M. et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. The New England journal of medicine 366, 883–892 (2012).
Reindl, W., Bowen, B. P., Balamotis, M. A., Green, J. E. & Northen, T. R. Multivariate analysis of a 3D mass spectral image for examining tissue heterogeneity. Integrative biology : quantitative biosciences from nano to macro 3, 460–467 (2011).
Woo, H. K., Northen, T. R., Yanes, O. & Siuzdak, G. Nanostructure-initiator mass spectrometry: a protocol for preparing and applying NIMS surfaces for high-sensitivity mass analysis. Nature protocols 3, 1341–1349 (2008).
Rockwood, A. L. & VanOrden, S. L. Ultrahigh-speed calculation of isotope distributions. Anal Chem 68, 2027–2030 (1996).
Rockwood, A. L., Vanorden, S. L. & Smith, R. D. Rapid Calculation of Isotope Distributions. Anal Chem 67, 2699–2704 (1995).
Lawson, C. L. & Hanson, R. J. Solving least squares problems. (Prentice-Hall, Englewood CliffsN. J., 1974).
Price, J. C. et al. Measurement of human plasma proteome dynamics with (2)H(2)O and liquid chromatography tandem mass spectrometry. Analytical biochemistry 420, 73–83 (2012).
Zeisel, S. H. Choline: an essential nutrient for humans. Nutrition 16, 669–671 (2000).
Acknowledgements
This work was supported by the Low Dose Radiation Program, Office of Biological and Environmental Research of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231 and by the National Institutes of Health through grant RC1NS069177. We would also like to thank M. Bissell and M. Balamotis for insightful comments, J. Gray, G. Karpen and R. Baran for valuable discussions and S. Kogan for guidance in histopathology analysis.
Author information
Authors and Affiliations
Contributions
T.N. and B.B. provided conception and overall experimental design. K.L., B.B. and T.N. analyzed data and wrote the manuscript. J.M., K.L., J.P. and Y.H. designed and performed experiments and contributed to manuscript editing. S.M. provided histopathology analysis of stained tumor sections. M.H. advised on isotopic analysis, clinical application of deuterium enrichment and contributed to manuscript editing.
Ethics declarations
Competing interests
Dr. Marc Hellerstein receives compensation as a member of the scientific advisory board of KineMed, Inc. and owns stock in the company. Dr. John Price is an employee of KineMed. Dr. Marc Hellerstein, Dr. Trent Northen, Dr. Benjamin Bowen and Dr. Katherine Louie have applied for a patent related to this technology. Dr. Jian-hua Mao, Yurong Huang and Dr. Stephanie McAlhany declare no potential conflict of interest.
Electronic supplementary material
Supplementary Information
Supplementary tables, figures and captions
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Louie, K., Bowen, B., McAlhany, S. et al. Mass spectrometry imaging for in situ kinetic histochemistry. Sci Rep 3, 1656 (2013). https://doi.org/10.1038/srep01656
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep01656
This article is cited by
-
Following de novo triglyceride dynamics in ovaries of Aedes aegypti during the previtellogenic stage
Scientific Reports (2021)
-
Applications of stable isotopes in MALDI imaging: current approaches and an eye on the future
Analytical and Bioanalytical Chemistry (2021)
-
Optical imaging of metabolic dynamics in animals
Nature Communications (2018)
-
Ultra-High Mass Resolution MALDI Imaging Mass Spectrometry of Proteins and Metabolites in a Mouse Model of Glioblastoma
Scientific Reports (2017)
-
Modern quantitative analytical tools and biosensors for functional studies of auxin
Journal of Plant Biology (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
