
Abstract
Networks of transcription factors (TFs) are thought to determine and maintain the identity of cells. Here we systematically repressed each of 100 TFs with shRNA and carried out global gene expression profiling in mouse embryonic stem (ES) cells. Unexpectedly, only the repression of a handful of TFs significantly affected transcriptomes, which changed in two directions/trajectories: one trajectory by the repression of either Pou5f1 or Sox2; the other trajectory by the repression of either Esrrb, Sall4, Nanog, or Tcfap4. The data suggest that the trajectories of gene expression change are already preconfigured by the gene regulatory network and roughly correspond to extraembryonic and embryonic fates of cell differentiation, respectively. These data also indicate the robustness of the pluripotency gene network, as the transient repression of most TFs did not alter the transcriptomes.
Similar content being viewed by others


Introduction
Transcription factors (TFs) bind to cognate DNA motifs in enhancers/promoters of target genes and regulate their expression. To decipher the structure and dynamics of gene regulatory networks, we used a strategy to monitor the effect of global transcriptome perturbation caused by the altered expression of single transcription factors (TFs). Previously we have focused on the gain-of-function assay, i.e., the overexpression of TFs and have carried out detailed whole-genome transcriptome analyses after inducing a total of 137 TFs one at a time1,2. As a complimentary approach, loss-of-function approaches have been used more often in cell biology. Indeed, there has been a prevailing paradigm that loss-of-function assays are a more effective way to reveal such a complex TF network. Although large-scale screening of cell phenotypes has been successfully carried out with libraries of shRNAs3,4, only a limited number of studies have performed global gene expression profiling5,6,7,8.
Results
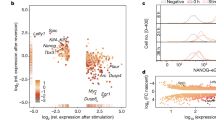
To assess the effectiveness of the loss-of-function approach, here we have repressed the expression of each TF with shRNAs, followed by transcriptome analysis with whole-genome expression microarrays (Fig. 1a). We selected 100 TFs based on their relevance to gene regulation1,2 and their relatively high expression9 in mouse ES cells (Fig. 1b). We developed and tested four independent shRNAs for their effectiveness in repressing the expression of each TF in ES cells. We used the best shRNAs for the first round of transcriptome analyses and the second best shRNAs for the second round as replications. For the first round, greater than 2-fold knockdown of TFs by gene-specific shRNAs was confirmed at the mRNA level for the majority of TFs (n = 89) (Fig. 1c, Supplementary Table 1) and at the protein level for all of 28 randomly selected TFs, including some that showed <2 fold reduction in RNA levels (Fig. 1d). If a TF was well repressed (>2 fold change of mRNA) by the first shRNA but this manipulation affected the expression of only a small number of downstream genes, we then assigned a low priority to this TF and did not use the second shRNA. For the majority of genes (n = 55), we carried out the second round of transcriptome analyses. Based on preliminary time-course experiments (Supplementary Figure 1), we chose to collect RNA samples 72 hours after the transient transfection of shRNAs, which permitted the detection of early changes in gene expression. The proportion of cells expressing shRNA was maximized by high transfection efficiency (~90%) and puromycin selection. The use of two independent shRNAs for each TF helped us to alleviate the major concern of shRNA experiments – off-target effects. Specificity of gene response was evident from the positive correlation between replications with different shRNA (Fig. 2) and from the enrichment of TF targets (genes with TF binding sites in promoters) within sets of genes that were upregulated or downregulated following the knockdown of TFs (Fig. 3). Results from the knockdown of Pou5f1 and Sox2 match well with published data on the downregulation of these genes in knockout cell lines with tetracycline-controlled transgenes (Supplementary Figure 2).

Knockdown of 100 transcription factors (TFs) in mouse ES cells with shRNA.
(a) experiment design; (b) list of TFs repressed in this study; TFs in bold were knocked down with two independent shRNAs; (c) change of mRNA expression of 100 TFs evaluated by microarrays and qPCR (see Supplementary Table 1); (d) Western blot shows the reduction of TF proteins; * - non-specific band.

Gene expression response after the knockdown of 16 trabscription factors with two different shRNA; r = correlation, logratio is log10; data are shown for 13929 genes with average log-expression >2.

Enrichment of transcription factor (TF) binding to genes that change their expression after the knockdown of TFs.
(a) gene rank plots for selected TFs: genes were sorted by the change of their expression and the proportion of targets was estimated in a sliding window of 500 genes; (b) z-value estimated using Parametric Analysis of Gene Set Enrichment (PAGE) which evaluates the enrichment of TF targets among genes that were upregulated or downregulated after the knockdown (see Supplementary Table 4 for the source of ChIP-seq/ChIP-chip data).
Surprisingly, the knockdown with shRNA of only a small subset of TFs (12 out of 100) was associated with a substantial transcriptome change (response of >100 genes) within 3 days after transfection (Fig. 4, Supplementary Tables 1, 2). In particular, it is interesting to note that the knockdown of Esrrb showed the most dramatic transcriptome changes, suggesting its importance in pluripotency. The majority of manipulated TFs (n = 67) showed very limited changes in gene expression profiles (<10 significant downstream genes). These results indicate that the TF-network that maintains the undifferentiated state of ES cells is surprisingly robust and is resistant to change caused by the abrupt loss of highly expressed TFs. This may be explained by the functional compensation of other factors; for example, the function of Klf4, Klf2 and Klf5 is known to be redundant in ES cells10. Alternatively, only a small number of TFs are indeed hubs of the network and form a core TF-network, whereas the majority of TFs, though expressed in undifferentiated ES cells, are in the periphery of the entire TF-network, governing only a small number of downstream target genes.

Knockdown of 100 transcription factors (TFs) with shRNA.
(a) Major TFs that changed transcriptome significantly after the knockdown (FDR < 0.05, change > = 2 fold) (see Supplementary Tables 1,2).(b) List of other TFs whose knockdown resulted in a change of expression of <20 genes.
To associate gene expression changes caused by the knockdown of TFs with the direction of cell state change, we carried out principal component analysis (PCA) for expression profiles of 2666 genes that showed a significant change of expression based on ANOVA (FDR < 0.05) (Fig. 5a). The PCA revealed that there were mainly two directions of state changes from an undifferentiated state in ES cells. A change in one direction (tentatively called, “trajectory 1”) was caused by the knockdown of either Pou5f1 or Sox2. A change in the other direction (tentatively called, “trajectory 2”) was caused by the knockdown of either Esrrb, Sall4, Nanog, or Tcfap4. Gene expression changes specific to each trajectories were also evident from the heatmap (Fig. 5b, Supplementary Table 3). The transition through trajectory 1 was associated with the upregulation of 655 genes, which include many trophectoderm-related genes (Cdx2, Ascl2, Cebpa, Esx1, Hand1 and Rhox6: Group A), indicating the extraembryonic direction of differentiation and the downregulation of 489 genes (Group B). By contrast, the transition through trajectory 2 was associated the upregulation of 287 genes, which include many epiblast-related genes inferred from the expression profiles of epiblast stem cells11 (Fgf5, En1, Sox6, Lrp2, Irx1, Lhx1, Msx1, Hoxa2: Group C), indicating the embryonic direction of differentiation and the downregulation of 167 genes (Group D). In addition, 294 genes were upregulated and 154 genes were downregulated following both transitions through trajectory 1 and 2 (Groups E and F, respectively).

Transcriptome changes of ES cells after the knockdown of transcription factors (TFs).
(a) principal components analysis (PCA) of log expression change of 2666 significant genes after the knockdown of transcription factors; (b) heatmap that shows gene expression changes associated with the Trajectory 1 (i.e., knockdown of Pou5f1 or Sox2) and Trajectory 2 (i.e., knockdown of Esrrb, Sall4, Nanog and Tcfap4) (see Supplementary Table 3).
The fact that removing either Pou5f1 or Sox2 had similar transcriptome change (a state change through the trajectory 1) is reasonable, as Pou5f1 and Sox2 are known to form dimers and bind to the promoters of target genes12. However, it was unexpected that repressing any of Esrrb, Sall4, Nanog, or Tcfap4 caused a similar transcriptome change (a state change through trajectory 2), which was distinct from the change caused by repressing either Pou5f1 or Sox2. Similar to the situation with trajectory 1, it is possible that Esrrb, Sall4, Nanog and Tcfap4 interact13 and regulate the same set of target genes. Alternatively, these TFs are a part of a distinct TF subnetwork, which together regulates the downstream target genes.
To gain further insights into these issues, we examined if the TFs bind to the promoter region of genes whose expression was changed by these TFs by assessing the enrichment of targets (i.e., genes with binding sites of TFs) among genes that change their expression in a trajectory-specific way (Groups A–D, Fig. 5b). Among 100 TFs examined in the current work, we used the information of TF-binding sites by ChIP-seq analyses that were publicly available for 28 TFs (Supplementary Table 4) and compared their binding prevalence to promoters/enhancers of upregulated versus downregulated genes. We found that genes associated with each trajectory differed substantially in the kind of TFs that bind to their promoter regions (Fig. 6): the trajectory 1 was characterized by the enrichment of targets of Pou5f1, Sox2, Nanog, Btbd14b and Tcf3 in downregulated genes (Group B); the trajectory 2 was characterized by the enrichment of targets of Esrrb and Klf4 in downregulated genes (Group D) and the enrichment of targets of Polycomb TFs (Eed, Suz12, Phc1, Jarid2 and Mtf2) and Trp53 in upregulated genes (Group C). Taken together, these results indicate that genes associated with the trajectory 1 and 2 are regulated by different sets of TFs.

Proportion of targets of various transcription factors (TFs) (i.e., genes with TF binding sites) among genes that change their expression following trajectories 1 (a) and 2(b); (c) z-value that compares the proportions of targets of various TFs between gene groups that were upregulated versus downregulated (see Supplementary Tables 3, 4); dashed line – significance level (p = 0.05).
Only those TFs which had a significant difference in binding to upregulated genes versus downregulated genes are shown.
Discussion
The data presented here highlight the presence of internal constraints in gene regulatory networks, which allow two main ways out of the pluripotent state specific for ES cells, something depicted as “Creod” by Conrad Waddington14. These data also indicate the robustness of the pluripotency TF network, as the perturbation caused by the transient repression of most TFs did not alter the transcriptomes. Considering that the repression of only a handful of TFs cause the exit of ES cells from the undifferentiated state, these TFs are most likely key TFs that prevent ES cells from cell differentiation. As summarized in Fig. 7, the expression of Pou5f1 and Sox2 prevent ES cells from differentiating through the trajectory 1, which seems to correspond to trophectoderm differentiation; whereas the expression of Esrrb, Sall4, Nanog and Tcfap4 prevent ES cells from differentiating through the trajectory 2, which seems to correspond to embryonic differentiation.

Two trajectories of short-term transcriptome change in ES cells following manipulation of transcription factors (TFs).
Trajectory 1 (extraembryonic direction) is characterized by the block of Pou5f1 and/or Sox2 signaling (via downregulation or interference), which leads to the downregulation of targets of Pou5f1 and Sox2. Trajectory 2 (embryonic direction) is characterized by the downregulation of targets of Esrrb and Klf4 and upregulation of targets of Polycomb factors while Pou5f1 and Sox2 are still active.
Among TFs that preferentially bind to genes downregulated along the trajectory 1 (group B in Fig. 5b), only Pou5f1 and Sox2 actively prevent extraembryonic differentiation, because the transcriptome of cells changed along trajectory 1 after the knockdown of these two TFs, but not after the knockdown of Nanog, Btbd14b and Tcf3 (Fig. 5a). Although binding sites of Nanog mostly coincide with binding sites of Pou5f1 and Sox215,16, only 86 targets of Pou5f1 and/or Sox2 are downregulated after the knockdown of Nanog and cells do not differentiate towards trophectoderm. Nanog-dependent targets of Pou5f1 and Sox2 included a large number of germline and testis-related genes (Prdm1, Prdm14, Kit, Esrrb, Notch4, Inhbb, Spats1, Zyg11a, Scgb3a1, Ifitm1, Sfrp1, Hck, Ttpa, Morc1), which is consistent with the idea that Nanog is associated with germline competence17. Initial report on the trophectoderm differentiation of mouse ES cells following the knockdown of Nanog5 was not confirmed by later studies7,18,19.
Among TFs that preferentially bind to the genes downregulated along the trajectory 2 (group D in Fig. 5b), only Esrrb actively prevents gene expression change based on our knockdown experiment (Fig. 7). Although targets of Klf4 are also over-represented within group D, the knockdown of Klf4 did not cause transcriptome change along trajectory 2. The lack of effect of Klf4 can be explained by its replacement by other TFs of the same gene family (Klf2 and Klf5), which can occupy the same binding sites10. Published gene expression data from a triple knockdown of Klf4, Klf2 and Klf5 indicates that ES cells undergo differentiation towards embryonic lineages (i.e., upregulation of epiblast-related Fgf5, Acta1, Irx5, Msx1 and no upregulation of trophectoderm-related Cdx2, Esx1, Hand1, Plac1 and Dlx3 on days 2 and 4)10. The importance of Klf4 for sustaining the undifferentiated state of ES cells is consistent with its ability to revert epiblast stem cells to the ground pluripotent state20. Thus, Esrrb and the KLF family of TFs are likely to control the change of gene expression along the trajectory 2, while core pluripotency TFs (Pou5f1 and Sox2) are still active (Fig. 7). The importance of Esrrb in ES cell differentiation is further supported by the fact that it is a positively-regulated target of Nanog and Sall4, as follows from our results on gene expression change after the knockdown of Nanog and Sall4 combined with published genome-wide mapping of TF binding sites16. Because the knockdown of Nanog and Sall4 caused gene expression change along trajectory 2, we can hypothesize that the effect of these TFs is mediated by downregulation of Esrrb. Also, Esrrb is known to be strongly downregulated in epiblast stem cells11; its knockdown causes ES cell differentiation towards epiblast-like cells5; and it regulates ES cell self-renewal via Gsk3/Tcf3 pathway21.
The mechanisms that upregulate targets of Polycomb factors along trajectory 2 are not clear. None of the Polycomb factors is downregulated substantially to explain this effect. Moreover, the knockdown of each Polycomb TF did not cause much change in the expression of downstream genes (Fig. 4, Supplementary Table 2). This is consistent with the finding that disruption of PRC1 and PRC2 complexes does not affect self-renewal of ES cells22. It is possible that Esrrb, Klf4, or their downstream targets, directly recruit or stabilize Polycomb factors, but there is no sufficient experimental evidence to support such a mechanism.
The strategy of systematic knockdown of TFs followed by gene expression profiling with microarrays allows a higher level of understanding of the causative relationship between TFs and their target genes, because alternative approaches (e.g., phenotype screening or analyzing correlations between gene expression profiles and TF binding to promoters) do not differentiate between causation and mere correlation. The systematic knockdown of 100 TFs reported here is the largest project of this kind in mouse ES cells. These data sets are substantial addition to our previous studies, in which a total 137 TFs are overexpressed in mouse ES cells, individually, followed by the global gene expression profilings1,2. The preferred direction of perturbation of TFs depends on the level of their expression in a given cell type: knockdown is generally more informative for TFs with high expression, whereas induction should be used for silent and low-expressed TFs. Especially, the use of the standard ES cell culture condition in our current and past studies1,2 has merits in terms of data comparability for the compendium analysis. However, this could also limit the generalization of the conclusions drawn from the current data sets and the use of a variety of culture conditions in the future is desirable. For example, it is known that Esrrb is necessary and sufficient to mediate self-renewal downstream of Gsk3 inhibition in LIF + 2i media, but it is dispensable for the self-renewal in LIF + FBS media21. Changing culture conditions would certainly add new dimensions to gene regulatory networks and open new differentiation pathways. Another limitation of transient shRNA-based strategy is that it considers only immediate effects in gene expression following the manipulation of TFs and thus, our results do not cover long-term effects related to self-renewal and differentiation as previously reported17,21. Nonetheless, the global inference of gene regulatory networks in ES cells in the standard culture condition can facilitate methods for their controlled differentiation into various cell types for therapeutic purposes.
Methods
Sequences for 4 shRNA were designed to target the 3′ untranslated region of genes and 2 of them were selected (Supplementary Table 5) based on the strong repression effect and minimal expression change in a housekeeping gene (Tuba1a) and gene associated with antiviral response (Oas1a). ES cells, MC1R (passage 20) established previously1 were cultured without feeder cells in standard media. After expansion, these cells were co-transfected with 1.6 μg of shRNA expression vector and cultured with puromycin starting at 24 h after transfection. Cells were harvested 72 h after transfection. We selected 72 h time point as a trade-off between two objectives: (a) to capture early responses of target genes and minimize indirect responses and (b) to observe robust changes in gene expression. The early time point (48 h) was not chosen due to relatively weak responses: only 111 genes changed their expression by >5-fold, as compared to 256 and 579 genes at 72 h and 96 h, respectively. The late time point (96 h) was not chosen, because it may include the response of many non-target genes that are affected indirectly. In particular, almost 1/2 of genes (48.3%) that changed their expression by >2-fold at 96 h did not respond at earlier time points (Supplementary Figure 1). The proportion of GFP-positive cells in culture, measured by flow cytometry, was high enough (>74% for all shRNA) to justify using all cells for gene expression profiling. We chose not to use sorted cells because sorting often results in cell damage followed by the induction of stress-related genes. RNA was extracted with Trizol™ (Invitrogen). Efficiency of knockdown was quantified by real time qPCR, microarrays and Western blot (Fig. 1c, d); primers and antibodies are listed in Supplementary Tables 6, 7. Gene expression was profiled with NIA Mouse 44 K Microarray v3.0, Agilent Technologies, #01508723. The data was batch-normalization for compatibility of experiments. Microarray data are submitted to GEO/NCBI (GSE26520). Two shRNA were used for the majority of TFs and we treated them as biological replications using ANOVA. Some of these experiments included also 2 technical replications where cells were split after transfection cultured separately and RNA was hybridized to separate microarrays. Further details on cell manipulation and data analysis can be found in the project website: http://esbank.nia.nih.gov/. For statistical analysis we used NIA Array Analysis, which estimates the False Discovery Rate (FDR) to account for multiple hypothesis testing24. All genes shown as significant in Fig. 4 and Supplementary Table S2 satisfy stringent statistical criteria (FDR < 0.05, fold change >2). Response of genes to the knockdown of TFs was measured as a logratio (i.e., difference between mean log-transformed expression in the knockdown experiment and global median expression). Principal component analysis (PCA) of log-transformed gene expression was done using genes that showed statistically significant change in their expression (FDR < 0.05) with SVD method applied to the covariance matrix of log-transformed gene expression values within the NIA Array Analysis.
Published ChIP-seq data on binding of 28 TFs (Supplementary Table 4) was compiled from either provided lists of gene symbols, or genome locations of binding sites. We used a distance of 15 Kb from the TSS as a generic threshold for associating binding sites with specific genes. TFs associated with ES cell pluripotency are often co-localized on DNA, making multiple transcription factor-binding loci (MTLs)16. Because these loci are associated with the binding of at least 4 TFs, they are highly reliable and can be used for detecting enhancers located far from the TSS. We used MTLs to add information on enhancers located within 200 Kb from the TSS for Pou5f1, Sox2, Nanog, Esrrb, Tclcp2l1, Klf4, Stat3 and Sall4 (Supplementary Table 4). For the TFs that have multiple sets of targets in our database, we selected the data that yielded highest enrichment scores (PAGE, see below) for sets of genes affected by manipulation of TFs, or combined multiple data sets.
Enrichment of target genes in subsets of genes that are upregulated and/or downregulated following the manipulation of a TF is quantified using a modified Parametric Analysis of Gene Enrichment (PAGE)25. PAGE is based on the comparison of the average expression change in a specific subset of genes, xset, with the average expression change in all genes, xall:

where nset is the size of the gene set and SDall is standard deviation of expression change among all genes. We modified this method by applying equation (1) to the subset of N top upregulated and another subset of N top downregulated genes rather than to all genes combined, which allowed us to detect enrichment of the same gene set among both upregulated and downregulated genes. The value of N = 5000 was selected experimentally because it appeared that enrichment of genes with TF binding sites is always limited to the top 5000 upregulated or downregulated genes. The probability distribution of expression change within subsets of N upregulated and downregulated genes is not normal; however, because we compare averages for large sets of genes (usually, nset is >50), the probability distribution of these averages is close to normal based on the central limit theorem26. Thus, it is reasonable to use equation (1) as approximation.
For the heatmap (Fig. 5b), we selected 2046 named genes (excluding olfactory receptors and Riken clones) with >1.5 fold median change following the knockdown of Pou5f1 and Sox2 (trajectory 1) or knockdown of Esrrb, Sall4, Nanog and Tcfap4 (trajectory 2). These genes were then classified into 6 response groups as follows: if the logratio of gene expression change was >2 fold greater following one trajectory that another trajectory, then the gene was considered specific to the former trajectory. A small number of genes (N = 18) responded in opposite directions following trajectories 1 and 2. We classified them according to the strongest absolute value of the median logratio of gene expression change. Within each group, genes were sorted using hierarchical clustering, TMEV, ver 3.127.
References
Nishiyama, A. et al. Uncovering early response of gene regulatory networks in ES cells by systematic induction of transcription factors. Cell Stem Cell (2009).
Correa-Cerro, L. S. et al. Generation of mouse ES cell lines engineered for the forced induction of transcription factors. Sci Rep 1, 167 (2011).
Hu, G. et al. A genome-wide RNAi screen identifies a new transcriptional module required for self-renewal. Genes Dev 23, 837–48 (2009).
Chia, N. Y. et al. A genome-wide RNAi screen reveals determinants of human embryonic stem cell identity. Nature 468, 316–20 (2010).
Ivanova, N. et al. Dissecting self-renewal in stem cells with RNA interference. Nature 442, 533–8 (2006).
Amit, I. et al. Unbiased reconstruction of a mammalian transcriptional network mediating pathogen responses. Science 326, 257–63 (2009).
Guttman, M. et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 477, 295–300 (2011).
Hurley, D. et al. Gene network inference and visualization tools for biologists: application to new human transcriptome datasets. Nucleic Acids Res 40, 2377–2398.
Cloonan, N. et al. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat Methods 5, 613–9 (2008).
Jiang, J. et al. A core Klf circuitry regulates self-renewal of embryonic stem cells. Nat Cell Biol 10, 353–60 (2008).
Tesar, P. J. et al. New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature 448, 196–9 (2007).
Remenyi, A. et al. Crystal structure of a POU/HMG/DNA ternary complex suggests differential assembly of Oct4 and Sox2 on two enhancers. Genes Dev 17, 2048–59 (2003).
van den Berg, D. L. et al. An Oct4-centered protein interaction network in embryonic stem cells. Cell Stem Cell 6, 369–81 (2010).
Waddington, C. H. The strategy of the genes (George Allen & Unwin Ltd, London, 1957).
Boyer, L. A. et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 122, 947–56 (2005).
Chen, X. et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 133, 1106–17 (2008).
Chambers, I. et al. Nanog safeguards pluripotency and mediates germline development. Nature 450, 1230–4 (2007).
Liu, N. et al. Identification of genes regulated by nanog which is involved in ES cells pluripotency and early differentiation. J Cell Biochem 104, 2348–62 (2008).
Loh, Y. H. et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat Genet 38, 431–40 (2006).
Guo, G. et al. Klf4 reverts developmentally programmed restriction of ground state pluripotency. Development 136, 1063–9 (2009).
Martello, G. et al. Esrrb is a pivotal target of the Gsk3/Tcf3 axis regulating embryonic stem cell self-renewal. Cell Stem Cell 11, 491–504 (2012).
Leeb, M. et al. Polycomb complexes act redundantly to repress genomic repeats and genes. Genes Dev 24, 265–76 (2010).
Carter, M. G. et al. Transcript copy number estimation using a mouse whole-genome oligonucleotide microarray. Genome Biol 6, R61 (2005).
Sharov, A. A., Dudekula, D. B. & Ko, M. S. A web-based tool for principal component and significance analysis of microarray data. Bioinformatics 21, 2548–9 (2005).
Kim, S. Y. & Volsky, D. J. PAGE: parametric analysis of gene set enrichment. BMC Bioinformatics 6, 144 (2005).
Rice, J. A. Mathematical statistics and data analysis, 1 v. (various pagings) (Thomson/Brooks/Cole, Belmont, CA, 2007).
Saeed, A. I. et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques 34, 374–8 (2003).
Acknowledgements
This research was supported in part by the Intramural Research Program of the NIH, National Institute on Aging.
Author information
Authors and Affiliations
Contributions
All authors participated in designing the study, carrying out experiments, analyzing the results, interpreting the data, or participating in the discussion. AAS and MSHK wrote the manuscript text and prepared figures. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Table 2
Supplementary Information
Supplementary Table 3
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Nishiyama, A., Sharov, A., Piao, Y. et al. Systematic repression of transcription factors reveals limited patterns of gene expression changes in ES cells. Sci Rep 3, 1390 (2013). https://doi.org/10.1038/srep01390
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep01390
This article is cited by
-
Atlas of regulated target genes of transcription factors (ART-TF) in human ES cells
BMC Bioinformatics (2022)
-
Metabolic control of DNA methylation in naive pluripotent cells
Nature Genetics (2021)
-
Perturbation-based gene regulatory network inference to unravel oncogenic mechanisms
Scientific Reports (2020)
-
The antiproliferative ELF2 isoform, ELF2B, induces apoptosis in vitro and perturbs early lymphocytic development in vivo
Journal of Hematology & Oncology (2017)
-
Generation and gene expression profiling of 48 transcription-factor-inducible mouse embryonic stem cell lines
Scientific Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
