
Abstract
Production of renewable commodity chemicals from bio-oil derived from fast pyrolysis of biomass has received considerable interests, but hindered by the presence of innumerable components in bio-oil. In present work, we proposed and experimentally demonstrated an innovative approach combining atmospheric distillation of bio-oil with co-pyrolysis for mass production of renewable chemicals from biomass, in which no waste was produced. It was estimated that 51.86 wt.% of distillate just containing dozens of separable organic components could be recovered using this approach. Ten protogenetic and three epigenetic compounds in distillate were qualitatively identified by gas chromatography/mass spectrometry and quantified by gas chromatography. Among them, the recovery efficiencies of acetic acid, propanoic acid and furfural were all higher than 80 wt.%. Formation pathways of the distillate components in this process were explored. This work opens up a fascinating prospect for mass production of chemical feedstock from waste biomass.
Similar content being viewed by others

Introduction
The past century has witnessed the huge success of petrochemical industry, but the rapid depletion of petroleum reserves and increased global concern on the sustainability of chemical industry have motivated substantial research on conversion of renewable and carbon-neutral biomass energy into marketable chemicals. Fast pyrolysis is a scalable thermochemical technology, which can depolymerize lignocellulosic biomass in a single oxygen-starved reactor at a very short reaction time and moderate temperature (673–873 K)1,2. The liquid product from fast pyrolysis, known as bio-oil, is a storable and transportable oxygenated biofuel and can be directly used for heat and electricity generation in static burners3,4. Particularly, further processing bio-oil to transportation fuels and value-added chemicals has attracted tremendous interests in the past decade5,6,7.
The production of commodity and/or specialty chemicals from bio-oil is attractive, but it is also faced with several challenges. Unlike petroleum oil, bio-oil is a very complex mixture of water, carboxylic acids, aldehydes, ketones, alcohols, esters and anhydrosugars, to name a few. Hundreds of organic molecules have been identified in bio-oil8. The organic fraction in bio-oil is characterized by the wide distribution of polarity and molecular weight, which poses challenges to the efficient separation. Even worse, bio-oil is produced under thermodynamically non-equilibrium conditions, thus some oxygenated components in bio-oil tend to react with each other (e.g., polymerization, condensation, esterification and etherification) at room or higher temperatures, leading to pronounced thermal and storage instability of bio-oil9,10. To date, many techniques, such as catalytic steam reforming11, aqueous phase processing2,12, have been developed to convert bio-oil and/or its fractions into specific chemicals. Direct processing the raw bio-oil catalytically seemed to be unfavorable because severe coke deposition causes a rapid deactivation of catalysts at temperature over 353 K13. Several methods have been proposed to break the micro-emulsion of raw bio-oil, e.g., adding inorganic salts14 or a sufficient amount of water15,16 at first, then the resulting two phases could be further treated to obtain respective target products. Such a pre-treatment simplifies the problem to some extent. Vispute et al.7,12 reported that hydrogen, C1–C6 alkanes, polyols and chemical feedstocks (aromatics and olefins) could be produced through water extraction by adding a large amount of water into bio-oil (the volume of water quadrupled that of bio-oil) and effectively hydrogenating the aqueous phase, but they could not resolve the catalyst deactivation problem. Furthermore, the economic feasibility of such a process is disputable because of the low concentration of organics in water and the requirement for external hydrogen supply.
Conventional atmospheric distillation and rectification are a simple, mature and extensively used separation technique in petrochemical refinery. To date, it is also the only economically feasible and scalable pathway to separate the complex bio-oil mixture into chemical fractions in biorefinery. However, two serious problems have hindered research works, which has resulted in almost no publications in this area in the past two decades: 1) the production of difficult-to-handle residues (atmospheric distillation residue, ADR) by aging of bio-oil at high temperatures; and 2) the formation of unpredictable chemicals in distillation process. Hence, tremendous efforts have been invested to mitigate the aforementioned problems. Molecular distillation17 and reduced pressure distillation required a lower distillation temperature, but they merely reduced the occurrence of ADR and aging reaction, rather than to eliminate it. Whereas atmospheric distillation of bio-oil in a high-boiling-point alcohol18,19, e.g., n-butanol and glycerol, was reported to obtain chemicals without producing ADR, but dose of solvent drastically increased the costs.
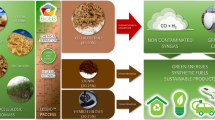
In the present work, an approach of atmospheric distillation of bio-oil without adding any reagent was proposed to obtain a distillate, which could be used as chemical feedstock (Fig. 1). We had no intention to avoid aging and coking of bio-oil as usual, but made full use of the ADR by circulating co-pyrolysis of rice husk and this ADR in an integrated system (see Fig. 1 and Supplementary Fig. S1). In such a system, no waste was discharged. The objectives of this work were: 1) to propose and experimentally verify a zero-waste process through combining atmospheric distillation with co-pyrolysis; 2) to obtain the stable chemical fractions in which the amount of compounds was decreased drastically; 3) to qualitatively identify and quantify the components and calculate the recovery efficiencies in fractions of distillate; and 4) to investigate the formation pathways of the distillated components.

Schematic of the direct atmospheric distillation coupled with co-pyrolysis process to produce chemical feedstock.
Results
The biomass was pyrolyzed to produce bio-oil containing hundreds of components. The bio-oil was distillated to obtain fractions of distillate and then all the ADR, which was concurrently produced along with distillate, was co-pyrolyzed with biomass. No waste was discharged from this integrated system. The integrated process is illustrated in Fig. S1 (Supplementary Information). The co-pyrolysis experiments using feedstock composed of ADR and rice husk at different ratios were carried out and the results indicated that the bio-oil after co-pyrolysis had no detectable change in yield (Fig. S2) and chemical composition (Table S1 and Fig. S3). This implies that the integrated process could resume all the produced ADR in atmospheric distillation.
The major compounds of bio-oil were qualitatively identified by gas chromatography/mass spectrometry (GC/MS) (Table S1). The main constituents of the organic fraction of bio-oil were commonly found carboxylic acids (e.g., formic acid, acetic acid, propanoic acid and n-hexadecanoic acid), furan derivatives (e.g., furfural, furfuryl alcohol and hydroxymethylfurfural), phenolic compounds (e.g., phenols and guaiacols), anhydrosugars (mainly levoglucosan), cylcopentanones and other carbonyl compounds. To obtain different compositions of distillates, the fractions of distillate were gathered at different temperatures. Figure 2 shows that the color of the distillate fractions became deepened with the increase in distillation temperature, except for the Fraction VI. The color of all fractions was much lighter than that of the raw bio-oil. Accumulated distillate accounting for 51.86 wt.% of the raw bio-oil was obtained (Fig. 3a) and its chemical composition was analyzed using GC/MS (see Supplementary Fig. S5a for the GC/MS chromatogram and Supplementary Table S5 for major identified compounds). Then, the concentrations of 13 major compounds in the distillate fractions were quantified using gas chromatography-flame ionization detector (GC-FID) (Fig. 3b). The organic fraction of distillate was mainly composed of light oxygenated compounds, whose boiling points range from 353 to 521 K. It is worth mentioning that the distillate had a potential to be further separated into pure chemicals because of the difference in their boiling points.

The photographs of raw bio-oil, different fractions of distillate and the atmospheric distillation residue.

(a) The cumulative distribution of distillates at different heating temperatures; (b) a typical GC chromatogram for quantification of components in distillate.
Most compounds in distillate (e.g., phenols, guaiacols, furan derivatives, acetic acid, propanoic acid and acetol) were commonly observed in bio-oils, suggesting that they were physically distilled off. Common compounds in Fractions I–VI were quantified and the results are shown in Figure 4. The water content of the distillate fractions decreased with the increase in temperature except for Fraction VI. This is because that most of water was evaporated at the first boiling point of bio-oil and the water content in the remaining oil gradually decreased. After the temperature reached 513 K or higher, some compounds reacted with each other to generate water and caused the increase in water content in Fraction VI. The concentrations of higher boiling-point compounds in the distillate fractions, such as p-cresol, o-cresol, guaiacol, phenol and furfuryl alcohol, increased with the increasing heating temperature, whereas those of the lower boiling-point compounds (e.g., n-butyric acid, furfuryl, propanoic acid, acetol and acetic acid) had a substantial decrease in Fraction VI. This implies that at such a high temperature these compounds were involved in the reaction of water generation (Figure 4a). Figure 4b shows that the yields of the distillated components based on the weight of the raw bio-oil. Higher recovery efficiencies were obtained by means of atmospheric distillation for acetic acid, propanoic acid and furfural, which reached 88.34 wt.%, 91.80 wt.% and 85.11 wt.%, respectively. The separation efficiencies of phenolic compounds were comparatively lower, attributed to their instability in the heating process, whereas the acids, alcohols and aldehydes exhibited an obvious instability only at higher temperature (>513K). This indicates that atmospheric distillation of bio-oil is a more favorable process for acids, alcohols and aldehydes.

(a) The distribution of organic compounds in distillate fractions; (b) the distribution of organic compounds in distillate and raw bio-oil and their separation efficiency in distillation.
Discussion
Figure 5 shows different plausible pathways for the conversion of lignocellulosic biomass into the value-added chemicals in distillate. Cellulose, hemicelluloses and lignin are the three main organic constituents of rice husk20,21. Considering the aromatic nature of lignin, phenols and guaiacols shown in Figure 5a were obviously the depolymerization products of lignin in the fast pyrolysis process and they were further physically separated in the atmospheric distillation and were present in the distillate. Light oxygenated compounds, e.g., acetic acid and acetol, were reported to be formed in fast pyrolysis process, both in the ring scission reaction of carbohydrates22,23 and the depolymerization of lignin24,25. Furfural, one of the most important platform chemicals in biorefinery, can also be obtained from the pyrolysis of cellulose, hemicelluloses and lignin25. The pyrolytic mechanism and primary reaction pathway of cellulose are still in debate despite of extensive studies26,27,28,29,30,31,32 on this topic. Levoglucosan (1,6-anhydro-β-D-glucopyranose), an anhydrosugar resulting from the glucosidic bond cleavage, has been recognized as an important intermediate in the thermal decomposion of cellulose. Primary thermal decomposition of cellulose may involve either the initial decomposition to levoglucosan followed by the subsequent dehydration, retroaldol condensation and fragmentation reactions of levoglucosan30 or depolymerization of cellulose or other intermediates, which competes with the formation of levoglucosan26,31. Furan derivatives, cyclopentenones, together with other aliphatic oxygenates could be obtained through decomposition of cellulose, dehydration of levoglucosan and both pathways and subsequent secondary reactions in the pyrolysis reactor.

(a) Formation pathway of molecules in distillate; (b) plausible chemical reactions in the atmospheric distillation of bio-oil.
The direct atmospheric distillation of bio-oil is not only a physical separation process, but also involves various chemical reactions between bio-oil components. In other words, the direct atmospheric distillation of bio-oil, although without adding any reagent, can also be viewed as “reactive distillation”. Three pieces of evidence can support this conclusion. Firstly, the coke formation indicates the occurrence of reactions, which increase molecular weight and carbon chain length. Secondly, water was produced in the distillation process. Taking the distillation of the raw bio-oil as an example, the water content and yield of the distillate of original bio-oil were measured to be 71.59 and 51.86 wt.%, respectively. There was 37.13 wt.% (51.86% × 71.59%) of water in distillate, which is higher than the initial water content (30.3 wt.%) in the raw bio-oil. This phenomenon clearly indicates that several dehydration reactions occurred to some extent in the atmospheric distillation. Lastly, several compounds, i.e., 2,3-butanedione, 2-butanone, 2,3-pentanedione, 3-penten-2-one, 1-acetoxy-2-butanone and 1-(2-furanyl)-ethanone, were found in distillate, but not detected in the bio-oil. Thus, they were reckoned as newly formed compounds in the distillation process.
Some reactions that possibly occurred in the distillation process are illustrated in Figure 5b. 2-butanone might be formed from the ketonic decarboxylation reaction of acetic acid and propanoic acid (Reaction (1) in Fig. 5b)33. The trace amount of inorganic minerals and entrained char fines34 in bio-oil might have an important catalytic effect on this type of reaction. As for 1-acetoxy-2-butanone, it was reckoned as the product of esterification reaction of acetic acid and 1-hydroxy-2-butanone (Reaction (2) in Fig. 5b). The removal of water in the distillation process shifted the chemical equilibrium in favor of formation of ester and the acids in the complex bio-oil could also catalyze the esterification reaction. 2,3-butanedione was reported to be derived from the pyrolysis of levoglucosan35,36, here it might also come from levoglucosan decomposition in the distillation process. Likewise, 2,3-pentanedione and 3-penten-2-one were also detected in the pyrolysate of cellulose at pyrolysis temperatures of 773–923 K37. Thus, it is reasonable to speculate that they were derived from the decomposition of levoglucosan and/or other anhydrosugar derivatives, which were intermediates in cellulose pyrolysis and present in bio-oil in the distillation process (Reactions (3–5) in Fig. 5b). Three types of newly formed compounds were quantified (Fig. 6). The concentrations of 2,3-butanedione, 2-butanone and 2,3-pentanedione reached their maximum content in the distillate Fractions III, V and IV, respectively. The formation of the new compounds in distillate indicates that the atmospheric distillation is a reactive process and is different from the traditional distillation.

(a) The concentration of 2,3-butanedione in different fractions of distillate; (b) the concentration of 2-butanone in different fractions of distillate; (c) the concentration of 2,3-pentanedione in different fractions of distillate; (d) the yield of 2,3-butanedione, 2-butanone and 2,3-pentanedione in the distillation.
In summary, a direct atmospheric distillation coupled with co-pyrolysis was demonstrated as a zero-waste approach for production of chemical feedstock. Compared with bio-oil, the amount of constituents of distillate was drastically decreased and could be further separated. Since the yield of bio-oil in biomass pyrolysis process is higher than 50.0 wt.%, about 10 wt.% of chemical feedstock by weight of feeding rice husk can be produced (see Supplementary Fig. S13) and it could be further separated into pure fractions based on their different boiling points. This facile process may be the most simple and economical approach to convert solid lignocellulosic biomass to chemicals at a large scale so far.
Methods
Materials
The rice husk used throughout the study was kindly provided by Anhui Yineng Bio-energy Co., China and collected from local rice mills. The rice husk was milled and screened and the portion with a particle size below 180 mesh (88 μm) was collected for the subsequent studies. Prior to the fast pyrolysis experiment, the samples were dried in an oven at 378 K for 12 h to remove moisture. The resulting dry rice husk particles were collected for the subsequent fast pyrolysis and analysis.
Atmospheric distillation of raw bio-oil
The atmospheric distillation was carried out in a round-bottom flask placed in an oil bath, in which a weighed amount of bio-oil was slowly heated to ~513 K under vigorous magnetic stirring. In this batch-mode distillation, volatiles flew upwards and bio-oil was continually condensed with the elevation of temperature, leaving the residues increasingly viscous. The bath was kept at the highest temperature for 20 min before the round-bottom flask was moved out of the oil bath and cooled to room temperature. The viscous ADR turned out to be a black solid at room temperature. It was crushed, ground and sieved to below 60 mesh (300 μm) and vacuum-dried at 343 K for 12 h to partially remove residual water and kept in a desiccator for further use.
Fast co-pyrolysis
The raw bio-oil was produced by the fast pyrolysis of rice husk alone. The fast co-pyrolysis experiments with certain proportions of a mixture of rice husk and ADR (originating from the distillation of raw bio-oil) were conducted in a downdraft fixed-bed fast pyrolyzer under nitrogen atmosphere at 723 K38.
Prior to the pyrolysis reaction, feedstock sample (5.000 ± 0.002 g) was prepared by physically mixing rice husk and ADR at certain ratios. ADR content in feedstock was set to vary in a range of 0–35 wt.%, when it was co-pyrolyzed with rice husk. Considering the maximum yield of ADR, the ADR content in feedstock was not further increased.
The mixture was then placed in the feeder, the pyrolysis zone was heated to 723 K by an electric furnace and the pyrolysis reactor was swept by nitrogen gas at a flow rate of 0.4 L/min for 20 min to wipe out air. Then, the feedstock entered the pyrolysis zone for 1–2 s. The resultant pyrolysis vapors were swept downstream by nitrogen at a flow rate of 0.16 L/min, of which the condensable fraction was quenched by cold ethanol (263 K) to obtain bio-oil. When pyrolysis was finished, the reactor was moved out of the heater and the residual char therein was cooled to room temperature in the atmosphere of nitrogen. The bio-oil in the condenser and the char in the tubular reactor were weighed to estimate their yields. Each experiment was conducted twice to verify the results.
Characterization methods
Water contents of the raw bio-oil and the distillate were determined through Karl-Fischer titration using chloroform/methanol (3:1, v/v) as a solvent. The elemental compositions (C, H and N) of the ADR, rice husk, whole distillate and bio-oils were determined on a Vario EL cube elemental analyzer (Elementar Analysensysteme GmbH, Germany). As for the proximate analysis of dry rice husk and ADR, the ash content was measured using the gravimetric method prescribed in ASTM D 3174-04, the content of volatile matter was determined using a non-isothermal thermogravimetric (TG) method39 and the content of fixed carbon was calculated by difference. In the TG method, 3.0-8.0 mg of samples were heated in a thermogravimetric analyzer (TGA-Q5000, TA Co., USA) at the atmosphere of 25 mL/min of N2. The temperature was programmed from room temperature to 383 K at a rate of 10.0 K/min and held for 10 min before ramped to 1173 K at a rate of 25.0 K/min. Apart from the contents of volatile matter, thermal characteristics of rice husk and ADR were also obtained by the TG method.
GC/MS was used to analyze the organic components of bio-oils qualitatively and semi-quantitatively. The separation was made on a HP-5MS capillary column (30 m × 0.25 mm × 0.25 μm). High-purity Helium was used as the carrier gas and the gas flow rate was held constant at 1 mL/min. The GC oven temperature was held at 313 K for 5 min and programmed to ramp at 4 K/min to 453 K and then at 10 K/min to 523 K. The oven was kept at the final temperature for 13 min. The injector temperature was 553 K and an injection volume of 1 μL was adopted with the split ratio set as 20:1. The mass spectrometer was operated in full scan mode and its mass range was 20–500 atomic mass units. The identification of the chromatographic peaks was based on an automatic library search (NIST library version 2.0). The quantification of compounds in liquid products is described in the Supplementary Information.
References
Czernik, S. & Bridgwater, A. V. Overview of applications of biomass fast pyrolysis oil. Energy Fuels 18, 590–598 (2004).
Gaunt, J. L. & Lehmann, J. Energy balance and emissions associated with biochar sequestration and pyrolysis bioenergy production. Environ. Sci. Technol. 42, 4152–4158 (2008).
Bridgwater, A. V. Biomass fast pyrolysis. Therm. Sci. 8, 21–50 (2004).
Singh, N. R., Delgass, W. N., Rebeiro, F. H. & Agrawal R. . Estimation of liquid fuel yields from biomass. Environ. Sci. Technol. 44, 5298–5305 (2010).
Huber, G. W., Iborra, S. & Corma, A. Synthesis of transportation fuels from biomass: chemistry, catalysts and engineering. Chem. Rev. 106, 4044–4098 (2006).
Mohan, D., Pittman, C. U. & Steele, P. H. Pyrolysis of wood/biomass for bio-oil: A critical review. Energy Fuels 20, 848–889 (2006).
Vispute, T. P., Zhang, H., Sanna, A., Xiao, R. & Huber, G. W. Renewable chemical commodity feedstocks from integrated catalytic processing of pyrolysis oils. Science 330, 1222–1227 (2010).
Mullen, C. A. & Boateng, A. A. Chemical composition of bio-oils produced by fast pyrolysis of two energy crops. Energy Fuels 22, 2104–2109 (2008).
Zhang, Q., Chang, J., Wang, T. & Xu, Y. Review of biomass pyrolysis oil properties and upgrading research. Energy Convers. Manage. 48, 87–92 (2007).
Diebold, J. P. A Review of the Chemical and Physical Mechanisms of the Storage Stability of Fast Pyrolysis Bio-oils. Report No. NREL/SR-570-27613, National Renewable Energy Laboratory, Golden, CO (2000
Wang, Z. et al. Production of hydrogen from catalytic steam reforming of bio-oil using C12A7-O--based catalysts. Appl. Catal., A 320, 24–34 (2007).
Vispute, T. P. & Huber, G. W. Production of hydrogen, alkanes and polyols by aqueous phase processing of wood-derived pyrolysis oils. Green Chem. 11, 1433–1445 (2009).
Huang, F., Li, W., Lu, Q. & Zhu, X. Homogeneous catalytic hydrogenation of bio-oil and related model aldehydes with RuCl2(PPh3)3 . Chem. Eng. Technol. 33, 2082–2088 (2010).
Song, Q., Nie, J., Ren, M. & Guo, Q. Effective phase separation of biomass pyrolysis oils by adding aqueous salt solutions. Energy Fuels 23, 3307–3312 (2009).
Sharma, R. K. & Bakhshi, N. N. Catalytic upgrading of pyrolysis oil. Energy Fuels 7, 306–314 (1993).
de Miguel Mercader, F. et al. Hydrodeoxygenation of pyrolysis oil fractions: process understanding and quality assessment through co-processing in refinery units. Energy Environ. Sci. 4, 985–997 (2011).
Wang, S. et al. Separation of bio-oil by molecular distillation. Fuel Process. Technol. 90, 738–745 (2009).
Mahfud, F. H., Melin-Cabrera, I., Manurung, R. & Heeres, H. J. Biomass to fuels: upgrading of flash pyrolysis oil by reactive distillation using a high boiling alcohol and acid catalysts. Process Saf. Environ. 85, 466–472 (2007).
Deng, L., Zhao, Y., Fu, Y. & Guo, Q. Green solvent for flash pyrolysis oil separation. Energy Fuels 23, 3337–3338 (2009).
Liou, T. H., Chang, F. W. & Lo, J. J. Pyrolysis kinetics of acid-leached rice husk. Ind. Eng. Chem. Res. 36, 568–573 (1997).
Teng, H., Lin, H. & Ho, J. Thermogravimetric analysis on global mass loss kinetics of rice hull pyrolysis. Ind. Eng. Chem. Res. 36, 3974–3977 (1997).
Patwardhan, P. R., Satrio, J. A., Brown, R. C. & Shanks, B. H. Product distribution from fast pyrolysis of glucose-based carbohydrates. J. Anal. Appl. Pyrolysis 86, 323–330 (2009).
Patwardhan, P. R., Brown, R. C. & Shanks, B. H. Product distribution from the fast pyrolysis of hemicellulose. ChemSusChem 4, 636–643 (2011).
Patwardhan, P. R., Brown, R. C. & Shanks, B. H. Understanding the fast pyrolysis of lignin. ChemSusChem 4, 1629–1636 (2011).
de Wild, P. Biomass Pyrolysis for Chemicals. Ph.D. Thesis., University of Groningen, Netherlands, (2011).
Ohnishi, A., Kato, K. & Takagi, E. Curie-point pyrolysis of cellulose. Polym. J. 7, 431–437 (1975).
Bradbury, G. W., Sakai, Y. & Shafizadeh, F. A kinetic model for pyrolysis of cellulose. J. Appl. Polym. Sci. 23, 3271–3280 (1979).
Pouwels, A. D., Eijkel, G. B. & Boon, J. J. Curie-point pyrolysis-capillary gas chromatography-high-resolution mass spectrometry of microcrystalline cellulose. J. Anal. Appl. Pyrolysis 14, 237–280 (1989).
Izawa, K., Matsukura, M. & Ishizu, Y. Curie-point pyrolysis of cellulose in the presence of potassium malate. Agric. Biol. Chem. 54, 957–963 (1990).
Lin, Y., Cho, J., Tompsett, G. A., Westmoreland, P. R. & Huber, G. W. Kinetics and mechanism of cellulose pyrolysis. J. Phys. Chem. C 113, 20097–20107 (2009).
Ranzi, E. et al. Chemical kinetics of biomass pyrolysis. Energy Fuels 22, 4292–4300 (2008).
Shen, D., Xiao, R., Gu, S. & Luo, K. The pyrolytic behavior of cellulose in lignocellulosic biomass: a review. RSC Adv. 1, 1641–1660 (2011).
Renz, M. Ketonization of carboxylic acids by decarboxylation: mechanism and scope. Eur. J. Org. Chem. 2005, 979–988 (2005).
Agblevor, F. A. & Besler, S. Inorganic compounds in biomass feedstocks. 1. Effect on the quality of fast pyrolysis oils. Energy Fuels 10, 293–298 (1996).
Shafizadeh, F. & Lai, Y. Z. Thermal degradation of 1,6-anhydro-.beta.-D-glucopyranose. J. Org. Chem. 37, 278–284 (1972).
Kawamoto, H., Murayama, M. & Saka, S. Pyrolysis behavior of levoglucosan as an intermediate in cellulose pyrolysis: polymerization into polysaccharide as a key reaction to carbonized product formation. J. Wood Sci. 49, 469–473 (2003).
Moldoveanu, S. C. Analytical Pyrolysis of Natural Organic Polymers. (Elsevier, Amsterdam, Netherlands, 1998).
Liu, W., Zeng, F., Jiang, H. & Yu, H. Total recovery of nitrogen and phosphorus from three wetland plants by fast pyrolysis technology. Bioresour. Technol. 102, 3471–3479 (2011).
Munir, S., Daood, S. S., Nimmo, W., Cunliffe, A. M. & Gibbs, B. M. Thermal analysis and devolatilization kinetics of cotton stalk, sugar cane bagasse and shea meal under nitrogen and air atmospheres. Bioresour. Technol. 100, 1413–1418 (2009).
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (50978242) and National 863 Program (2009AA06Z305).
Author information
Authors and Affiliations
Contributions
X.-S.Z. conducted most of experiments, analyzed the data and drafted the manuscript; G.-X.Y. assisted with the quantification of organic compounds in distillate fractions; H.J. conceived the experiments and supervised this project; W.-J.L. and H.-S.D. assisted with data analysis and co-pyrolysis experiments, respectively; all authors discussed the results and contributed to the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information-Copyrolysis
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Zhang, XS., Yang, GX., Jiang, H. et al. Mass production of chemicals from biomass-derived oil by directly atmospheric distillation coupled with co-pyrolysis. Sci Rep 3, 1120 (2013). https://doi.org/10.1038/srep01120
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep01120
This article is cited by
-
Physical and chemical characterizations of a reference e-cigarette used in animal testing
Scientific Reports (2023)
-
Preparation of high-crystalline and non-metal modified g-C3N4 for improving ultrasound-accelerated white-LED-light-driven photocatalytic performances
Scientific Reports (2023)
-
A Concise Review on the Synthesis, and Characterization of the Pyrolytic Lignocellulosic Biomass for Oil, Char and Gas Production: Recent Advances and its Environmental Application
Chemistry Africa (2023)
-
Highly Versatile Gum Acacia Based Swellable Microgels Encapsulating Cobalt Nanoparticles; An Approach to Rapid and Recoverable Environmental Nano-catalysis
Journal of Inorganic and Organometallic Polymers and Materials (2021)
-
Initial pyrolysis mechanism and product formation of cellulose: An Experimental and Density functional theory(DFT) study
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
