
Abstract
The molecular mechanisms underlying viral pathogenesis are yet poorly understood owed to the large number of factors involved and the complexity of their interactions. Could we identify a minimal set of host transcription factors (TF) whose misregulation would result in the transcriptional profile characteristic of infected cells in absence of the virus? How many of such sets exist? Are all orthogonal or share critical TFs involved in specific biological functions? We have developed a computational methodology that uses a quantitative model of the transcriptional regulatory network (TRN) of Arabidopsis thaliana to explore the landscape of all possible re-engineered TRNs whose transcriptomic profiles mimic those observed in infected plants. We found core sets containing between six and 34 TFs, depending on the virus, whose in silico knockout or overexpression in the TRN resulted in transcriptional profiles that minimally deviate from those observed in infected plants.
Similar content being viewed by others


Introduction
For over decades, plant molecular virology has been overly focused on the pathogen itself, studying their individual genes and products and their local effects on certain regulatory pathways related to antiviral responses or to symptoms development. The symptoms of viral infections result from the sum of direct effects (e.g., cytoplasmic accumulation of viral products or modifications in the cytoskeleton or in membrane structures) as well as of indirect effects from the alteration of host physiology, notably by diverting almost every cellular resource to the production of virus-specific components and by actively suppressing host defenses1,2. The arrival of genomic tools have allowed high-throughput genetic and metabolic screenings, providing unprecedented views of the plant host-virus interactions from a systemic perspective that would allow for a deeper understanding on how host and virus genotypes interplay in determining the pathological outcome of an infection3,4,5,6,7.
Microarray-based functional genomics, which provides a global view of transcriptional changes in host cells, has been the most commonly used method to study global changes during plant-virus interactions2,8,9,10,11,12,13,14,15,16. As a response to infection, hosts compensate by over- or under-expressing certain cellular pathways and deploying specific antiviral measures. Collectively, these alterations determine the type and strength of symptoms displayed by infected organisms as well as the virulence of the infection. Imposing the measured transcriptional changes in a biological network context, it was confirmed that host cells undergo a significant reprogramming of their transcriptome during infection17,18, which is possibly a central requirement for the mounting of host defenses. Moreover, Rodrigo et al. uncovered a general mode of plant virus action in which perturbations preferentially affect genes that are highly connected, central and organized in modules19, a mechanism of action that has been pervasively described for animal viruses20,21,22,23,24,25,26.
Inspired by an integrated computational-experimental approach for discovering genes and pathways that are targets of specific compounds27, herein, we aimed to computationally re-design the transcriptional regulatory network (TRN) of Arabidopsis thaliana by altering key transcription factors (TFs) in order to mimic the transcriptional response observed upon infecting the plant with several different virus. We will accomplish this goal by re-designing optimal genetic network using as starting point a genome-scale TRN model of the plant28. Hence, those computational re-designs will provide new insights about mechanisms related with virus-target interactions in the plant. Recently, many groups have proposed and implemented different approaches for genome-wide re-design, by knocking out and over-expressing genes, of prokaryotes and eukaryotes to control global gene expression29,30,31,32. Following this synthetic biology strategy, herein we have computationally re-designed A. thaliana TRN by exhaustively exploring multiple gene perturbations in the form of gene knockouts or over-expressions. Hence, we have corroborated in silico that several genetic modifications imposed on a critical set of TFs generates a high diversity in the transcriptome of the plant.
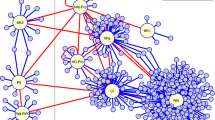
Could a reduced set of perturbed TFs mimic the plant's transcriptional response to viral infections? It is of outmost importance to harness the ability of using computational design to predict and optimize à la carte synthetic genomes with desired transcriptional responses (Figure 1). To address this question, we have developed an algorithm that uses as starting point a wild-type transcription regulation model, inferred from high-throughput microarray data28. This TRN is evolved using a heuristic optimization method that at each stage computes the updated gene expression profile and compares it with the one observed during viral infection. With this approach, we explored the space of re-engineered TRNs to find the optimal global network whose predicted transcriptional profile has a minimal distance to the one characteristic of viral infections. Consequently, the use of genomic techniques to develop design-guided models and the application of reverse-engineering methods, open the doors for delineating a high-resolution picture of host-pathogen interactions.

Schematic representation of the methodology followed for this study.
(A) Reverse-engineering to reveal gene sub-networks differentially altered by viral infection. (B) Reprogramming cells to mimic the plant transcriptomic responses observed upon viral infection by using computational genome redesign.
Results
We have developed a methodology to automatically re-design the TRN of A. thaliana to mimic the transcriptomic changes induced by perturbations. In particular, we have focused on the perturbations induced by the infection with a set of eight different viruses. For that, we hypothesized that symptoms of viral infections could be recreated in absence of the pathogenic agent by altering a minimal core set of TFs (Figure 1B). We used a genome-wide model of A. thaliana gene transcription based on ordinary differential equations (ODEs) to predict changes in gene expression after modifications such as TF knockout or overexpression28. This model contains 21929 genes (~82% of all non-redundant genes in A. thaliana), 1187 of which are putative TFs (~75% of all A. thaliana TFs) and consequently, are the potential candidates to be perturbed. Figure 2 shows a schematic representation of the evolutionary algorithm implemented, in which single gene mutations were made and then selected according to the fitness functions discussed in the Methods section. This algorithm explores the landscape of all possible TRN produced by over-expressing or knocking out A. thaliana TFs. Operationally, these perturbations were introduced by modifying the corresponding ODEs. At each evolutionary step, a population of such perturbed TRNs was generated and their corresponding expression profiles computed and compared with the target transcriptomic profile observed for each viral infection. Those TRNs showing the better matching with the target were selected for the next round of optimization, as thoroughly discussed in the Methods section. Fifty independent runs of this evolutionary optimization were generated to evaluate the convergence of the results.

Computational methodology.
Schematic representation of the algorithm used to predict optimal redesigns of the A. thaliana TRN that mimics the alterations induced by viral infections on the plant transcriptome.
Re-engineered TRNs that mimic the transcriptomic response characteristic of different viral infections
Figure 3 illustrates that re-engineered TRNs actually show expression profiles that are significantly closer to those observed in infected plants than the profile inferred for non-infected plants (wild-type). Different panels correspond to the optimizations done for the eight different viruses using the scoring method that takes into consideration only perturbations in TFs (Figure 2). The ordinates axis shows the computed expression score (Sexp). A perfect matching between the observed TRN and that computed for the re-engineered genome would have Sexp = 0. The larger the departure from zero, the poorer the matching between observed and predicted expression profiles. The black vertical line corresponds to the score computed for the wild-type TRN (i.e., non-infected plants). In blue, we show the observed distribution of scores after multiple optimization runs. In all cases, the optimization results in artificial TRNs that are closer to the observed transcriptome than to the wild-type. The best fit was obtained for TRV (Sexp < 1.96%; P < 0.001) and the worst for PPV (Sexp < 9.51%; P < 0.001). As an additional quality control, we also ran the optimization algorithm but without the selective constraint imposed by matching the observed transcriptional profiles (red bars in Figure 3). In these cases, the distributions of Sexp did not show any improvement but, instead, had average values larger than those observed for the wild-type TRN. As we would expect, considering not only TFs in the scoring function but all genes, predictability is not significantly improved, ranging between Sexp < 1.88% (P < 0.001) for CaLCuV and Sexp < 10.36% (P < 0.001) for PPV (Figure S1). Indeed, the Sexp obtained only with TFs or with all genes are highly correlated (Figure S2; r = 0.714, 6 d.f., P = 0.046), thus suggesting that the conclusion is robust to the choice of genes to be perturbed during the optimization process. Interestingly, this conclusion does not specifically hold for infection with TCV (Figure S2). For this virus, using only TFs results in a poor optimization. To better illustrate the matching between the gene expression of the re-engineered TRNs and those of the corresponding virus-infected plants, Figure 4 shows the TFs expression values for the different predicted TRNs. Hence, most of re-designed TRNs guarantee TFs expression patterns highly similar to those measured in the virus-infected plants and quite different from those observed in the non-infected plants.

Distributions of expression scores, Sexp.
Histogram of the scores obtained in 50 optimization processes (blue bars) forced to mimic the plant transcriptome changes observed after infection with eight different viruses. In the optimization process, only TFs were considered in the scoring function (S). Random simulations were computed without imposing any selective pressure (red bars). Black line shows the score obtained using the transcriptional model inferred of A. thaliana.

Heat maps comparing the expression profiles of TFs.
Expression profiles of the TFs measured in the infected plants and predicted in the best-optimized and random transcriptomes. Note that under- and over-expressed genes are plotted in red and green colors, respectively.
A minimal set of transcriptional factors guarantees TRN re-designs that mimic viral infections
Transcriptomic studies have shown that the number of A. thaliana TFs altered upon viral infection varies among different viruses, with TRV altering 11 and TEV-At17 altering 101 (gray bars in the upper panel of Figure 5A)4,19. What would be the number of TFs whose expression has been altered in the re-engineered TRNs? To answer this question we simply counted the number of the transcriptional modification done on each designed TRN. Figure 5A shows this information for each virus, both for TRNs evolved with the scoring function that only accounts for changes in TFs (blue bars) and for the scoring function that accounts for alterations in all genes (red bars). Surprisingly, after optimization, the number of altered TFs necessary to mimic viral infections was quite reduced for all viruses. For example, looking only at TRNs designed using the first scoring function, the minimum number of perturbations necessary was found for CaLCuV infection (6.40) and the maximum for PPV (34.24) (Figure 5A). Narrower ranges (from 1.92 to 9.50) were obtained for all viruses when the second scoring function was used instead.

Statistics of the number of TFs perturbed and of the type of perturbation.
(A) Number of TFs to perturb (knockout/over-expression) proposed by the optimization process in which only TFs or all genes are considered in the scoring function (blue and red bars, respectively, in the upper panel). Grey bars show the number of TFs differentially expressed that were identified by gene expression upon infection with eight different viruses. We also show the intersection between the perturbed TFs proposed in the design and those with differentially altered gene expression (bottom panel). Random selections of TFs for designing simulating optimization processes without selective pressure were computed to test statistical significance (*** P < 0.001). (B) Percentages of over-expressed and (C) knockout TFs proposed to be perturbed in each design for the eight viruses. Error bars show SD from the 50 simulations.
The set of TFs proposed by the design algorithm do not necessarily include all those whose expression has been observed in real infections (Figure 5A, lower panel). In general, a bootstrap test shows that the intersections between the lists of proposed TFs and observed altered TFs were not significantly enriched except in two instances. In the case of TEV-At17 40.15% of the proposed TFs were altered in the real infection, being the proposed set significantly enriched in observed TFs (Figure 5A lower panel; bootstrap test, P < 0.001). This significant enrichment does not exist when all genes were considered in the scoring function. Analogously, 18.73% of the proposed TFs perturbed in the redesigned TRNs were contained in the list of observed altered TFs for TRV (Figure 5A, lower panel; bootstrap test, P < 0.001) and this result remains significant independently of the gene set weighted by the scoring function.
Next, we observed that the lists of genes proposed by all re-designs were significantly enriched in biological functions related with response to biotic and abiotic stresses and in developmental processes (summarized in Additional file 1). This enrichment corroborates that the pathological outcome of viral infection can be reproduced in silico in absence of the viral agent by altering the appropriated plant cellular programs. Hence, the plasticity of A. thaliana transcriptome to generate specific expression pattern as a response to multiple genetic perturbations suggests that non-infected cells could easily mimic transcriptomic responses to diverse viral infections. What kinds of perturbations contribute the most to this plasticity? Figures 5B and 5C show, respectively, the percentages of over-expressed and knocked out TFs for each redesigned TRNs (as before, blue and red bars correspond to scoring functions that use only TFs or all genes). Overall, re-engineered TRNs included more gene knockouts than over-expressions for all viruses except for the re-designs mimicking PPV infection, which are more balanced, indicating that gene knockouts generate more plasticity in gene expression than overexpressing genes.
We have just shown that the list of TFs altered in the re-engineered TRNs were not necessarily the same set that have been shown to have altered expression in the real infections. Now, we want to explore whether this proposed set of TFs are located in the wild-type TRN close to TFs that are altered during real infections, as described in Figure 6A. If this is the case, then it can be argued that the proposed set of TFs does affect exactly the same target genes that may be affected by the TFs altered during viral infection. Otherwise, it can be argued that the proposed set of TFs mimics infection by completely different mechanisms. To evaluate this question we proceeded as follows. For each proposed TF we evaluated a topological parameter, ΦTF, which takes into account the minimum undirected shortest path between this TF and all the TFs significantly altered by viral infection (see Methods). The parameter ΦTF takes the value one if the proposed TF is included in the list of TFs altered in real infections and tends to zero as the distance to the closest altered TF increases in the network. Figures 6B and 6C show ΦTF for each virus (panel B corresponds to scoring functions using only TFs and panel C to all genes). The statistical significance of ΦTF was evaluated by generating random lists of transcription factors. Only TFs proposed in all re-designs of TuMV and TRV were significantly closer in the wild-type TRN to TFs differentially altered by viral infection (Figures 6B and 6C), for both scoring functions, than expected by sheer chance.

Measures of transcriptional information.
(A) Scheme to illustrate the topological properties in a context of the A. thaliana TRN between the perturbed TFs proposed by our methodology and those identified differentially altered under viral conditions. (B, C) Transcriptional information of all TFs proposed to be perturbed in the designs. Note that blue and red bars show TFs proposed by using our design methodology evaluating only TFs or all genes in the scoring function, respectively. Random undirected shortest paths were computed to evaluate the statistical significance of the topological distance between the TFs for designing with respect TFs selected randomly in the A. thaliana interactome (*** P < 0.001). Error bars show SD from the 50 simulations.
All together, these results confirm that the re-engineered TRNs proposed by our computational methodology mimic the transcriptomic response observed under real viral infections by altering a smaller and different set of targets than those observed during the transcriptomic characterization of real infections. Indeed, in general, they are not even neighbors in the wild-type TRN.
The number of proposed TFs to be perturbed and their connectivity degree correlates with the magnitude of the alterations in gene expression observed upon viral infection
The microarray data characterizing the infection of A. thaliana with each of the eight viruses shows significant variation in the number of genes whose expression was altered. Therefore, we hypothesized that in order to match the transcriptomic consequences of infection, our optimization algorithm shall propose more TF alterations for viruses that exert a large impact on the host transcriptome than for viruses that have a mild impact. To test this hypothesis we proceeded as follows. First, to collapse into a single quantity the impact of viral infection in the host transcriptome, we computed two different statistical measures, Pearson's correlation coefficient and the mutual information (MI) between the transcriptomic profiles of non-infected and infected plants. For viruses having minor impact in the plant transcriptome, the Pearson's coefficient will be close to one and the MI large. By contrast, a small correlation coefficient and low MI will reflect a strong perturbation in the plant's transcriptome. Second, we sought for associations between these indexes and the number of predicted altered TFs for each virus or their outgoing connectivity in the TRN. If our hypothesis is correct, we must observe significant negative correlations in these relationships. The largest changes in the transcriptome were found in cells infected by PPV, whereas the action of the CaLCuV showed the smallest variation in the host expression profile. Figures 7A and 7B show the expected negative and significant correlations between the number of proposed alterations, using the scoring function based only in TFs and the overall impact of the viral infection measured as the Pearson's correlation (Figure 7A: r = −0.890, 6 d.f., P = 0.003) and as the MI (Figure 7B: r = −0.800, 6 d.f., P = 0.017). Analogously, Figures 7C and 7D also illustrate a significant negative correlation between the average outgoing connectivity of the proposed TFs and the overall impact of the viral infection measured as Pearson's correlation (Figure 7C: r = −0.789, 6 d.f., P = 0.019) and as MI (Figure 7D: r = −0.927, 6 d.f., P = 0.001). Additionally, we compared the average outgoing connectivity for the proposed TFs for each virus with respect to the average of all TFs of A. thaliana TRN (Figure 7G). Only in the case of PPV, the proposed TFs regulate significantly more genes than expected from random samples in the TRN. We could hypothesize that an optimal way to perturb the global transcriptome, thus mimicking the virus effect would be to alter transcriptional hubs.

Complexity of the proposed solutions.
(A, B) Relationship between the number of TFs proposed by the design algorithm to be perturbed, (C, D) their connectivity degree and (E, F) a combination of both variables and the correlation (evaluated as Pearson correlation (A, C and E) or mutual information (B, D and F)) between the gene expression profiles of the wild-type plants and plants infected with each of the eight viruses. Only TFs were considered in the scoring function during the optimization process. (G) Average of the outgoing connectivity for TFs proposed for each virus. Note that average connectivity of all TFs of A. thaliana (TRN) was compared with respect to each virus to compute statistical differences (*** P < 0.05). Error bars show SD for the 50 simulations in (A, B) and SEM in (C, G).
Next, we hypothesized a relationship between a new parameter, which collapses the two previous characteristics of the proposed TFs into a single figure, ΨV and the overall impact of the viral infection. Interestingly, we computed high and significant correlations by using the Pearson's correlation (Figure 7E: r = −0.936, 6 d.f., P = 0.001) or the MI (Figure 7F: r = −0.900, 6 d.f., P = 0.002) as measurements of impact of the viral infection in transcriptome.
Hence, we could conclude that the amount and type (connectivity degree) of proposed perturbations needed to mimic a viral infection is directly dependent on the overall effect that the virus exerts on the host transcriptome: the larger the effect, the more perturbations are needed and vice versa.
A crucial set of TFs is pervasively proposed in the redesigned TRNs
For each virus, we have run 50 independent optimization processes. Do all of them represent completely different solutions? Or by contrast, do all of them contain a preferred set of TFs? If the second situation is true, then we can suggest that the over-represented TFs represent a critical set of highly relevant TFs that, eventually, may be the focus of future experimental validation.
For each virus, we tabulated the 50 lists of proposed TFs and calculated the probability of finding each TF in these lists. This probability can be taken as a measure of the design robustness of the prediction for each TF. A TF with a very low design robustness (e.g., < 25/50) means that it may have very low relevance in mimicking the transcriptomic profile induced by the virus. By contrast, a TF with high design robustness (e.g., > 25/50) will be indicative that such TF plays a central role in mimicking the transcriptomic symptoms. Figure 8 (blue bars) shows such degree of design robustness for perturbations of all TFs for the eight viruses. In this case the scoring function used in the optimization process only accounted for TFs. The different simulations did not share most of the TFs proposed. However, a certain number of TFs were common for more than half of the simulation. This number of crucial TFs varies among viruses, ranging from zero (TRV) to 19 (PPV). Analogously, Figure S3 also shows the equivalent results but weighting all genes in the scoring function. In this case the number of TFs that we can identify as crucial is much lower: At1g43700 and At1g24625 (TMV), At1g24625 (TRV) and At1g17880 (TuMV) related to DNA mediated transformation, cellular response to sulfate starvation, negative regulation of cell differentiation, nuclear import, regulation of response to stress, sulfate transport and response to salt stress. As a way to evaluate the statistical significance of these results, we generated, for each virus, a new set of 50 simulations but without using the match in expression profile (Sexp) in the scoring function. The distribution of design robustness for these TRNs is shown on Figure 8 (red bars). In all cases, the distributions of design robustness did not reach values larger than 0.2, thus confirming that the existence of critical TFs could not be explained by chance. For each virus, the centrality and shape of both distributions were compared and found significantly different (Mann-Whitney test, P < 0.001; Kolgomorov-Smirnov test, P < 0.001). Table 1 shows the lists of all TFs defined as critical for each virus. Interestingly, among the 47 TFs proposed, 22 are involved in several developmental processes; nine in responses to biotic and abiotic stresses and the rest do not have been assigned to any specific function. This enrichment in TFs involved in development may be consistent with the symptoms induced upon viral infection, which in most cases involve dwarfism, leaf malformations and curling and delays in the emergence and development of inflorescences.

Histogram of the robustness of the TFs proposed by the design algorithm for the eight viruses (blue bars).
Only TFs were considered in the scoring function for designing. TFs with design robustness > 0.5 are described in Table 1. The red bar histograms correspond to the robustness of TFs selected from randomly re-engineered TRNs.
Proposed TFs common for different viruses
Finally, we explored the overlap between the lists of TFs whose expression was altered in the TRNs designed for the different viruses. For each virus, this list includes all the TFs proposed at least in one of the 50 simulations (i.e., not restricting the analyses to the critical set described in the above paragraph). As in previous sections, the lists of TFs were build both using the scoring functions based on TFs and in all genes. Figure 9A shows the number of TFs that are found in the lists of one or more viruses (blue and red bars corresponding to whether only TFs or all genes were accounted for in the scoring function). Specifically, around 700 TFs were virus-specific, but a large fraction (over 400) was shared by at least two viruses. Among these, ca. 200 TFs were shared by at least three viruses. In the right side of the distribution, we found 16 TFs shared by seven viruses. Finally, all viruses only share three TFs: At1g50640, At2g35940 and At2g37650. At1g50640 corresponds to the ethylene-responsive transcription factor 3 (ERF3) that negatively regulates the ethylene-mediated signaling pathway and gene transcription. At2g35940 corresponds to the BEL1-like homeodomain 1 protein (BLH1) that regulates transcription in response to abscisic acid stimulus. At2g37650 encodes for a TF of the GRAS family involved in root and leaf development and in the negative regulation of flower development. Hence, the number of TFs identified for an increasing number of viruses decreases exponentially (r = 0.969, 7 d.f., P < 0.0001), showing that there is a significant relationship between the number of TFs identified for one virus compared to another. In addition, since transcriptomics shows that the hosts tend to respond in a similar way to different pathogens, we identified a high number of TFs in common between multiple viruses with respect to list of TFs generated randomly (grey bars in Figure 9A). This would further support the proposal that TFs are not selected randomly and consequently, our methodology identified a specific set of TFs that mimic the common effects of virus infection.

Commonalities among viruses in the proposed TFs.
(A) Number of common TFs proposed to be perturbed by the model (blue and red bars) and by 1000 random simulations (grey bars) for several viruses. (B, C) Neighbor-joining dendograms obtained from the similarity matrix computed from overlapping lists of TFs proposed to be perturbed in the different designs for the eight viruses. Note that only TFs ((B) and blue bars in (A)) or all genes ((C) and red bars in (A)) were considered in the scoring function for designing.
The overlap between pairs of lists was further quantified using the similarity index 2nxy/(nx + ny), where nxy is the number of common entries in the two lists and nx and ny the length of each list. A similarity matrix containing all pairwise comparisons was constructed and used to build neighbor-joining dendograms that cluster together viruses according to the similarity of their proposed lists of TFs (Figure 9B and figure 9C, using the scoring function based only on TFs or considering all genes, respectively). Three groups result when the first scoring function was used (Figure 9B). The first group is formed by PPV, TEV and TCV, the second group by TRV, TuMV, CaLCuV and TEV-At17, while TMV appears as the most dissimilar virus. Given the nature of the data used to build up this dendogram (i.e., similarities among lists of TFs whose alteration mimic the symptoms of infection), not surprisingly, the clustering does not reflect phylogenetic relationships between viruses (PPV, TEV, TEV-At17 and TuMV are all classified within the same taxonomic genus, the Potyvirus) nor whether they share common hosts in nature (TCV, TuMV, CaLCuV and TEV-At17 infect either A. thaliana or related hosts in the Brassicaceae). By contrast, when the scoring function used in the optimization process takes into account all genes, the dendogram obtained clusters together all the A. thaliana-infecting viruses (Figure 9C). Thus suggesting that host-driven selection may have determined the set of genes that viruses infecting the same host manipulate to optimize their replication.
Discussion
The ultimate aim of synthetic biology is de novo construction of an organism by combining standardized parts33. More modest aims are to re-design already existing biological systems, by adding new elements or rewiring existing ones, to produce certain new metabolites or to improve certain physiological processes34,35. Despite being modest, these are very challenging goals that will clearly gain from the development of computational tools that will help to a rational in silico design of new circuits, allowing comparing them, analyzing their dynamical properties and selecting the optimal one to be finally implemented in vivo29,36. Here, we take a computational synthetic biology approach to tackle a somehow different and new problem: identifying sets of cellular factors whose perturbation results in the same transcriptomic pattern that is observed in plant cells infected by viruses. This approach is opposite to the usual research pathway taken by molecular virologists that consists in first identifying cell proteins that interact with viral proteins and that result in the typical symptoms of an infection8,17,18. Our approach compares the transcriptomic profiles of non-infected plants with those observed for virus-infected plants and searches in silico for those minimal sets of TFs or genes whose alteration (knockout or overexpression) results in a predicted transcriptomic pattern that minimally deviates from the observed in infected plants. We have performed this computational search for a set of eight plant viruses all infecting the same host, A. thaliana.
Plants have evolved defense mechanisms to recognize pathogens and defeat viruses, but viruses have developed elements that interfere and suppress these mechanisms. The computational methodology proposed in this article allows exploring the plasticity of A. thaliana transcriptome in response to the alteration of certain key TFs. Although our approach only proposed synthetic perturbations in TFs, it has been established that some viruses affect the expression and activity of enzymes. A good example of such alteration in non-TFs is the effect of geminivirus infection on the activity of kinases such as adenosine kinase37,38,39, SNF1-related kinases40 and shaggy-related kinase AtSKeta41. We have addressed the problem of re-design a plant TRN to mimic the transcriptomic response observed upon viral infection but in absence of any intracellular pathogen. In the case of eight different viruses, our methodology rendered re-engineered TRNs that, in silico, captured the transcriptional responses of the infected host A. thaliana. Interestingly, we found that for all eight viruses, the optimization process resulted in a significant improvement in the similarity between observed and simulated data. Surprisingly, this high degree of mimicking was reached by manipulating a reduced number of TFs associated to developmental processes and to responses to biotic and abiotic stresses. In other words, our method is not trivially identifying TFs that were already affected by the viruses but different sets of TFs whose coordinated alteration result in a similar transcriptomic pattern. Note that in the collected microarray experiments, many TFs are expressed at levels undistinguishable from the background. Thus, the minimal set of TFs proposed could be biased towards those expressed at higher levels. However, although this may certainly be a potential source of bias, we believe it is not of importance because (i) our lists would still represent a conservative set of TFs and (ii) TFs with low changes in expression across the microarray compendium may still be associated to high values of its corresponding regulatory effect parameters28 (see Methods section). Other potential source of bias comes from the fact that only approximately 75% of all A. thaliana TFs have been used in our study, those that appear annotated as such in TAIR, thus we may be missing some relevant ones. This deficiency may be solved in the future as annotations in databases are curated and improved.
Virus infections are dynamic processes and the reprogramming of the transcriptomic profile may undergo important changes along the curse of infection. Since the datasets used in this study correspond to samples taken at a single time point, the results may be somehow biased towards TFs affecting late-response genes rather than to those being altered at early stages of the infection and that, not unlikely, may correspond to defense-related genes. However, it is worth noting that in a previous meta-analysis of the same experimental data used here19, we failed to detect significant differences in gene expression when experiments were arbitrarily ranked according to the day in which samples were taken (i.e., early, intermediate and late). At face value, this result suggested that observed differences in gene expression among the eight viruses used in this study are not explicable by the time at which samples were taken. Nonetheless, as more transcriptomic data become available in public databases, especially data from temporal samples, we will address dynamic modeling of TRN rewiring.
As we expected, the complexity of the redesigned TRNs, in terms of number of necessary TF to be perturbed, correlated with the amount of change induced by each viral infection to the transcriptome of infected plants. In other words, the more differences exist between non-infected and infected plants, the more TFs need to be altered to minimize differences between both transcriptomic profiles. In addition, we found certain degree of overlap between the TFs selected in each run of the optimization algorithm, providing evidence that a set of essential TFs is able to generate high plasticity in gene expression of the plant. This is to say, these TFs are not trivially selected but instead are keystone in maximizing the similarity between simulated and observed transcriptomic profiles. Finally, we found that some TFs were shared among different viruses in a phylogenetic-independent manner. That is, viruses do not group according to their phylogenetic relatedness but according to the list of altered TFs in common. We interpret this observation as a consequence of the optimization process that identifies core TFs responsible for the big alterations in transcriptomic profiles.
Experimental validation of our computational predictions for each virus requires the generation of mutant A. thaliana lines with altered expression of the TFs listed in Table 1. This represents a considerable amount of work beyond the scope of this manuscript. However, for CaLCuV, TCV and TuMV our method has robustly suggested that the alteration of a single TF may be enough to mimic viral infection; giving some hope validating our predictions by exploring available information for these TFs. If alteration of a single TF translates into large transcriptomic and/or phenotypic effects, then that would at least confirm that our method correctly selects for changes potentially mimicking the consequences of viral infection. By contrast, if the effect of these proposed alterations is limited, then some warning flags should be raised against the value of our predictions. In the case of CaLCuV, At5g59820 encodes for a zinc finger protein involved in the response to several stress responses, notably to high light and cold acclimation. It is expressed in multiple tissues and at different stages of development. Overexpression of this TF has a cascade effect and alters the expression levels of other 24 cold-responsive genes, including C-repeat binding factor (CBF) genes42,43. In the case of TCV, the At1g59930 TF is involved in the regulation of expression of maternally imprinted genes44. Currently, ca. 50 imprinted A. thaliana genes are known. These genes encode for proteins that are implicated in a wide range of functions and processes, including other TFs, proteins involved in hormone signaling, components of the ubiquitin protein degradation pathway, regulators of histone and DNA methylation and small RNA pathway proteins44. Finally, At5g07690 predicted for TuMV encodes for an R2R3 MYB29 TF expressed at many developmental stages (including leaf senescence and petal differentiation and expansion) and in many different tissues. MYB29, together with MYB28, is involved in the methyl jasmonate-mediated induction of a set of aliphatic glucosinolates (GSLs) biosynthetic genes45,46. GSLs are well known for their role as signaling molecules in plant defense against herbivory, wounding, induction of systemic resistance, responses to other hormones and, overall, affect the adaptation of plants to variable environments47,48. In conclusion, even for the most simple situations described in Table 1, alteration of the predicted TFs results in a cascade of effects, thus confirming that our method is not predicting TFs at random but precisely those which are central for multiple processes.
This computational work, identifying reduced sets of TFs that result in mimicking the symptoms of infection, opens the doors to future experiments that may use the A. thaliana gene knock out collections not only to validate our prediction but also as a way of reach a better understanding of the molecular mechanisms of viral pathology. Even perhaps as potential targets for future therapeutic interventions. In this sense, the overexpression of one or several of the core TFs suggested by our study may result in tolerant plants, which will not develop symptoms despite being infected. Transgenic overexpression of these key TFs may compensate for the reduction in concentration forced by viral infections thus significantly reducing symptoms.
Methods
Transcriptional regulatory network of Arabidopsis thaliana
A linear model based on ODEs providing the steady states of each mRNA was used to describe transcriptional gene regulations28. Transcriptomic data were normalized and represented in log-scale. Thus, the mRNA steady state from the ith gene, gi, is given by

where αi is its constitutive transcription rate, βij is the regulatory effect that gene j has on gene i and δi is the degradation coefficient. Time was conveniently scaled such that δi = 1 and the model is assumed in steady state ( ). Notice that each gene expression is bounded in a range interval defined by the minimum (
). Notice that each gene expression is bounded in a range interval defined by the minimum ( ) and maximum (
) and maximum ( ) value of all its conditions obtained from the 1436 hybridization experiments collected in the TAIR website (www.arabidopsis.org). To calibrate TF expression, the newly redefined constitutive transcription rate
) value of all its conditions obtained from the 1436 hybridization experiments collected in the TAIR website (www.arabidopsis.org). To calibrate TF expression, the newly redefined constitutive transcription rate  included a perturbative term (εi) that fit only the TF expression profile (yopt) for a defined optimal condition in which cell growth is maximal.
included a perturbative term (εi) that fit only the TF expression profile (yopt) for a defined optimal condition in which cell growth is maximal.
Plant viruses and transcriptomic data
Seven positive-sense single-stranded RNA viruses and one virus whose genome is composed by a single-stranded circular ambisense DNA molecule all infecting a common plant host, A. thaliana, were selected. The set of RNA viruses comprises three members of the Potyviridae family, Tobacco etch potyvirus (TEV), Turnip mosaic potyvirus (TuMV) and Plum pox potyvirus (PPV), two members of the family Virgaviridae, Tobacco mosaic tobamovirus (TMV) and Tobacco rattle tobravirus (TRV) and one member of the Tombusviridae family, Turnip crinkle carmovirus (TCV). We also considered a laboratory- strain of TEV (TEV-At17) evolved in and adapted to A. thaliana10. The ssDNA virus included in the study was the member of the Geminiviridae family Cabbage leaf curl begomovirus (CaLCuV).
TEV and TEV-At17 expression data (two-color raw data, NCBI GEO accession GSE11088) were obtained from ecotype Ler-0 plants 14 days post-inoculation (dpi)9,10. TuMV data (Affymetrix raw data, ArrayExpress accession e-mexp-509) were obtained 5 dpi from ecotype Col-0 plants16. PPV data (Affymetrix preprocessed data, NCBI GEO accession GSE11217) were obtained 17 dpi from Col-0 plants12. TMV data (two-color raw data, deposited in www.bio.puc.cl/labs/arce/index.html) were obtained from ecotype Uk-4 plants 10 dpi14. TRV data (two-color raw data), NCBI GEO accession GSE15557, GSE155562 and GSE15558) were measured eight dpi from Col-0 leafs. TCV data (two-color raw data, NCBI GEO accession GSE29387) were quantified 10 dpi in Col-0 plants. Finally, CaLCuV data (Affymetrix raw data, ArrayExpress accession E-ATMX-34) were collected from Col-0 plants 12 dpi11. The list of differentially expressed genes was obtained by performing a fold-change criterion of z > 1.96 over all genes (averaging replicates). The effect of heterogeneity in the host ecotype used in different experiments (Ler-0 for TEV and TEV-At17, Uk-4 for TMV and Col-0 for the rest) and in the time at which samples were obtained (ranging from 5 to 17 dpi) has been explored else where19.
Genome-wide multiple-optimization
Our algorithm searches possible reconfigurations of the global transcription regulatory network such as that the expression profile of the re-engineered genome mimics the transcriptional response of the host infected by different viruses. We address this problem by using a high efficient heuristic optimization. We suggest genome reconfigurations that include multiple knockouts or over-expression among the 1187 TFs identified in the A. thaliana genome, by imposing in the model  or
or  , respectively, or both types of perturbations in order to enlarge the combinatorial space of perturbed genomes targeting the transcriptional response given under viral infection. We started from the inferred model and at each step in the optimization process, we randomly selected a new TF to evaluate its three states (knockout, over-expression or wild-type) and update the model with the best-scored scenario until all TFs have been manipulated.
, respectively, or both types of perturbations in order to enlarge the combinatorial space of perturbed genomes targeting the transcriptional response given under viral infection. We started from the inferred model and at each step in the optimization process, we randomly selected a new TF to evaluate its three states (knockout, over-expression or wild-type) and update the model with the best-scored scenario until all TFs have been manipulated.
We use a bi-objective scoring function, φ, to force a minimum average deviation to the viral infection expression profile in steady state (Sexp) with the minimum number of genetic perturbations (Spert),

where

and

ϑ is the parameter that defines the weighting factor of each objective function (ϑ = 0.5 for all simulations),  is the expression profile under viral infection of all genes (Ng) of A. thaliana and λg ∈ (0, 1) is a parameter defined for each gene that differences those genes differentially expressed in the microarray data measured under viral conditions. In fact, we can divide the objective function representing the expression score into two terms: a first that quantifies expression deviations from genes that have been identified as altered in the transcription and a second to compute the rest of genes. We imposed λg = 1 for minimizing the fact that genes differentially expressed in the microarrays could have more importance in Sexp. NTF is the total number of TFs annotated in A. thaliana and
is the expression profile under viral infection of all genes (Ng) of A. thaliana and λg ∈ (0, 1) is a parameter defined for each gene that differences those genes differentially expressed in the microarray data measured under viral conditions. In fact, we can divide the objective function representing the expression score into two terms: a first that quantifies expression deviations from genes that have been identified as altered in the transcription and a second to compute the rest of genes. We imposed λg = 1 for minimizing the fact that genes differentially expressed in the microarrays could have more importance in Sexp. NTF is the total number of TFs annotated in A. thaliana and  is the number of TFs that the model suggests to be perturbed.
is the number of TFs that the model suggests to be perturbed.
Transcriptional information
First, we computed the shortest paths  , in the plant TRN, between a TF (among the set of proposed perturbations in a given design) and a set of TFs potentially affected by global changes in gene expression, g (genes differentially altered by viral infection in the microarray). Hence, to measure the degree of contribution that a TF has to mimic a target gene expression of the TRN of A. thaliana, we defined a parameter
, in the plant TRN, between a TF (among the set of proposed perturbations in a given design) and a set of TFs potentially affected by global changes in gene expression, g (genes differentially altered by viral infection in the microarray). Hence, to measure the degree of contribution that a TF has to mimic a target gene expression of the TRN of A. thaliana, we defined a parameter

that evaluates the minimum shortest path (i.e., the minimal number of links between a TF and the genes differentially expressed under viral conditions).
Topological parameter of TRN designed
To characterize the impact of the set of TFs proposed in the TRN of A. thaliana, we defined a topological parameter for each viral infection, v,

that evaluates the number of TFs proposed,  and the average outgoing connectivity of such TFs,
and the average outgoing connectivity of such TFs,  , relative to the virus with maximum
, relative to the virus with maximum  or γ, respectively. Note that we fixed the weighting factor, λ = 0.5, to provide the same importance to both topological features.
or γ, respectively. Note that we fixed the weighting factor, λ = 0.5, to provide the same importance to both topological features.
References
Dodds, P. N. & Rathjen, J. P. Plant immunity: towards an integrated view of plant-pathogen interactions. Nat Rev Genet 11, 539–48 (2010).
Jenner, R. G. & Young, R. A. Insights into host responses against pathoghens from transcriptional profiling. Nat Rev Microbiol 3, 281–94 (2005).
Andeweg, A. C., Haagmans, B. L. & Osterhaus, A. D. Virogenomics: the virus-host interaction revisited. Curr Opin Microbiol 11, 461–6 (2008).
Elena, S. F., Carrera, J. & Rodrigo, G. A systems biology approach to the evolution of plant-virus interactions. Curr Opin Plant Biol 14, 372–7 (2011).
Friedel, C. C. & Haas, J. Virus-host interactomes and global models of virus-infected cells. Trends Microbiol 19, 501–8 (2011).
Peng, X. et al. Virus-host interactions: from systems biology to translational research. Curr Opin Microbiol 12, 432–8 (2009).
Tan, S. L., Ganji, G., Paeper, B., Proll, S. & Katze, M. G. Systems biology and the host response to viral infection. Nat Biotech 25, 1383–9 (2007).
Wise, R. P., Moscou, M. J., Bogdanove, A. J. & Whitham, S. A. Transcript profiling in host-pathogen interactions. Annu Rev Phytopathol 43, 329–69 (2007).
Agudelo-Romero, P. et al. Changes in the gene expression profile of Arabidopsis thaliana after infection with Tobacco etch virus. Virol J 5, 92 (2008).
Agudelo-Romero, P., Carbonell, P., Pérez-Amador, M. A. & Elena, S. F. Virus adaptation by manipulation of host's gene expression. PLoS ONE 3, e2397 (2008).
Ascencio-Ibáñez, J. et al. Global analysis of Arabidopsis gene expression uncovers a complex array of changes impacting pathogen response and cell cycle during geminivirus infection. Plant Physiol 148, 436–54 (2008).
Babu, M., Griffiths, J. S., Huang, T. S. & Wang, A. Altered gene expression changes in Arabidopsis leaf tissues and protoplasts in response to Plum pox virus infection. BMC Genomics 9, 325 (2008).
Espinoza, C., Medina, C., Somerville, S. & Arce-Jonhson, P. Senescence-associated genes induced during compatible viral interactions with grapevine and Arabidopsis. J Exp Bot 58, 3197–212 (2007).
Golem, S. & Culver, J. N. Tobacco mosaic virus induced alterations in the gene expression profile of Arabidopsis thaliana. Mol Plant-Microb Interact 16, 681–8 (2003).
Ishihara, T. et al. Comparative analysis of expressed sequence tags in resistant and susceptible ecotypes of Arabidopsis thaliana infected with Cucumber mosaic virus. Plant Cell Physiol 45, 470–80 (2004).
Yang, C. et al. Spatial analysis of Arabidopsis thaliana gene expression in response to Turnip mosaic virus infection. Mol Plant-Microb Interact 20, 358–70 (2007).
Whitham, S. A., Yang, C. & Goodin, M. M. Global impact: elucidating plant responses to viral infection. Mol Plant-Microb Interact 11, 1207–15 (2006).
Whitham, S. A. & Wang, Y. Roles for host factors in plant viral pathogenicity. Curr Opin Plant Biol 7, 365–71 (2004).
Rodrigo, G. et al. A meta-analysis reveals the commonalities and differences in Arabidopsis thaliana response to different viral pathogens. PLoS ONE 7, e40526 (2012).
Bushman, F. D. et al. Host cell factors in HIV replication: meta-analysis of genome-wide studies. PLoS Pathog 5, e1000437 (2009).
Calderwood, M. A. et al. Epstein-Barr virus and virus human protein interaction maps. Proc Natl Acad Sci USA 104, 7606–11 (2007).
De Chassey, B. et al. Hepatitis C virus infection protein network. Mol Syst Biol 4, 230 (2008).
MacPherson, J. I., Dickerson, J. E., Pinney, J. W. & Robertson, D. L. Patterns of HIV-1 protein interaction identify perturbed host-cellular subsystems. PLoS Comput Biol 6, e1000863 (2010).
Uetz, P. et al. Herpesviral protein networks and their interaction with the human proteome. Science 311, 239–42 (2006).
Watanabe, T., Watanabe, S. & Kawaoka, Y. Cellular networks involved in the influenza virus life cycle. Cell Host Microbe 7, 427–39 (2010).
Wuchty, S., Siwo, G. & Ferdig, M. T. Viral organization of human proteins. PLoS ONE 5, e11796 (2010).
Di Bernardo, D. et al. Chemogenomic profiling on a genome-wide scale using reverse-engineered gene networks. Nat Biotech 23, 377–83 (2005).
Carrera, J., Rodrigo, G., Jaramillo, A. & Elena, S. F. Reverse-engineering the Arabidopsis thaliana transcriptional network under changing environmental conditions. Genome Biol 10, R96 (2009).
Carrera, J., Elena, S. F. & Jaramillo, A. Computational design of genomic transcriptional networks with adaptation to varying environments. Proc Natl Acad Sci USA 109, 15277–82 (2012).
Segrè, D., Vitkup, D. & Church, G. M. Analysis of optimality in natural and perturbed metabolic networks. Proc Natl Acad Sci USA 99, 15112–7 (2002).
Burgard, A. P., Pharkya, P. & Maranas, C. D. OptKnock: A bilevel programming framework for identifying gene knockout strategies for microbial strain optimization. Biotechnol Bioeng 84, 647–57 (2003).
Isalan, M. et al. Evolvability and hierarchy in rewired bacterial gene networks. Nature 452, 840–5 (2008).
Kitney, R. & Freemont, P. Synthetic biology – the state of play. FEBS Lett 586, 2029–36 (2012).
Rodrigo, R., Carrera, J., Landrain, T. E. & Jaramillo, A. Perspectives on the automatic design of regulatory systems for synthetic biology. FEBS Lett 586, 2037–42 (2012).
Carrera, J. et al. Fine-tuning tomato agronomic properties by computational genome redesign. PLoS Comput Biol 8, e1002528 (2012).
Carrera, J., Rodrigo, G. & Jaramillo, A. Towards the automated engineering of a synthetic genome. Mol Biosyst 5, 733–43 (2009).
Wang, H., Hao, L., Shung, C. Y., Sunter, G. & Bisaro, D. M. Adenosine kinase is inactivated by geminivirus AL2 and L2 proteins. Plant Cell 15, 3020–32 (2003).
Wang, H., Buckley, K. J., Yang, X., Buchmann, R. C. & Bisaro, D. M. Adenosine kinase inhibition and suppression of RNA silencing by geminivirus AL2 and L2 proteins. J Virol 79, 7410–8 (2005).
Baliji, S., Lacatus, G. & Sunter, G. The interaction between pathogenicity proteins and adenosine kinase leads to increased expression of primary cytokinin-responsive genes. Virology 402, 238–47 (2010).
Shen, W. & Hanley-Bowdoin, L. Geminivirus infection up-regulates the expression of two Arabidopsis protein kinases related to yeast SNF1- and mammalian AMPK-activating kinases. Plant Physiol 142, 1642–55 (2006).
Piroux, N., Saunders, K., Page, A. & Stanley, J. Geminivirus pathogenicity protein C4 interacts with Arabidopsis thaliana shaggy-related protein kinase AtSKeta, a component of the brassinosteroid signaling pathway. Virology 362, 428–40 (2007).
Vogel, J. T., Zarka, D. G., Van Buskirk, H. A., Fowler, S. G. & Thomashow, M. F. Roles of the CBF2 and ZAT12 transcription factors in configuring the low temperature transcriptome of Arabidopsis. Plant J 41, 195–211 (2005).
Lee, C. M. & Thomashow, M. F. Photoperiodic regulation of the C-repeat binding factor (CBF) cold acclimation pathway and freezing tolerance in Arabidopsis thaliana. Proc Natl Acad Sci USA 109, 1504–9 (2012).
Hsieh, T. F. et al. Regulation of imprinted gene expression in Arabidopsis endosperm. Proc Natl Acad Sci USA 108, 1755–62 (2011).
Hirai, M. Y. et al. Omics-based identification of Arabidopsis Myb transcription factors regulating aliphatic glucosinolate biosynthesis. Proc Natl Acad Sci USA 104, 6478–83 (2007).
Sønderby, I. E., Burow, M., Rowe, H. C., Kliebenstein, D. J. & Halkier, B. A. A complex interplay of three R2R3 MYB transcription factors determines the profile of aliphatic glucosinolates in Arabidopsis. Plant Physiol 153, 348–63 (2010).
Sønderby, I. E., Geu-Flores, F. & Halkier, B. A. Biosynthesis of glucosinolates – gene discovery and beyond. Trends Plant Sci 15, 283–90 (2010).
Burow, M., Halkier, B. A. & Kliebenstein, D. J. Regulatory networks of glucosinolates shape Arabidopsis thaliana fitness. Curr Opin Plant Biol 13, 347–52 (2010).
Acknowledgements
We thank J.A. Daròs, M.A. Fares and G. Rodrigo for fruitful comments and suggestions and O. Voinnet and C. Llave for sharing with us the TCV and TRV transcriptomic data, respectively. This research was supported by grant BFU2009-06993 by the Spanish Secretaría de Estado de Investigación, Desarrollo e Innovación to S.F.E.
Author information
Authors and Affiliations
Contributions
J.C. and S.F.E. conceived and designed the study. J.C. performed the computational work. J.C. and S.F.E. analyzed the data and wrote the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary information
Supplementary Information
Supplementary Dataset 1
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Carrera, J., Elena, S. Computational design of host transcription-factors sets whose misregulation mimics the transcriptomic effect of viral infections. Sci Rep 2, 1006 (2012). https://doi.org/10.1038/srep01006
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep01006
This article is cited by
-
Identifying differentially coexpressed module during HIV disease progression: A multiobjective approach
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
