
Abstract
Gene targeting in embryonic stem cells (ESCs) has become the principal technology for generating knockout models. Although numerous studies have predicted that the disruption of p53 leads to increased developmental anomalies and malignancies, most p53 knockout mice develop normally. Therefore, the role of p53 in animal development was examined using rat knockout models. Conventionally generated homozygous KO males developed normally, whereas females rarely survived due to neural tube defects. Mutant chimeras generated via blastocyst injection with p53-null ESCs exhibited high rates of embryonic lethality in both sexes. This phenotype could be observed in one month by the use of zinc-finger nucleases. The p53-null ESCs were resistant to apoptosis and differentiation and exhibited severe chromosome instabilities in the chimera-contributed cells, suggesting an essential role for p53 in maintaining ESC quality and genomic integrity. These results demonstrate that p53 functions as a guardian of embryogenesis in the rats.
Similar content being viewed by others


Introduction
Over the past two decades, knockout (KO) technology in mice has helped to clarify the physiological function of a large number of genes. However, unexpected phenotypes have been observed in some cases, making it difficult to understand the role of the deleted gene, or to translate that data to the phenotypes of human diseases caused by mutations in such genes. Thus, gene-targeting techniques for other animals, such as rats, have long been sought. Many strategies for manipulating rat genes to generate loss-of-function models have been adapted from the mouse genetic toolbox, including conventional transgenesis by pronuclear injection1, RNA interference2, N-ethyl-N-nitrosourea (ENU) mutagenesis3,4 and transposon mutagenesis5,6,7. KO rats have been produced using Zinc-finger nuclease (ZFN) technology8,9, and, most recently, germline-competent rat ESCs and rat induced pluripotent stem cells (iPSCs) have been established by the addition of cell-signaling inhibitors to the culture medium10,11,12,13, making it possible to generate both transgenic (Tg)12,14 and KO rats15.
The tumor suppressor gene p53 is a good example of a gene whose function in mouse development requires further scrutiny. Donehower et al. first reported normal Mendelian ratios and postnatal development in p53 homozygous KO mice16. However, two other groups later showed that a fraction of homozygous KO females had fatal embryonic exencephaly, a defect in neural tube closure that results in an overgrowth of neural tissue in the midbrain region17,18. Such results indicate that, at least in some cases, p53 influences development in females19. In the case of p53 homozygous KO rats, neural tube defects (NTDs) in females were not found but increased susceptibility to tumor development was reported15,20,21.
p53 has been shown to regulate not only cell cycle arrest, apoptosis and DNA repair in many types of cells22, but also stemness, by suppressing Nanog expression in ESCs23. Considering these observations, malignant transformations may occur in p53-null ESCs and chimera development may be hindered. However, chimeric mutant mice have been successfully generated via the injection of blastocysts with iPSCs derived from p53-null mouse embryonic fibroblasts (MEFs) and germline transmission of the p53-null cells was also accomplished24,25. The properties of rat ESCs differ from those of mouse ESCs in that rat ESCs cannot be cultured in mouse ESC culture conditions due to their high sensitivity to differentiation signals26. Thus, an approach using p53-null rat ESCs might reveal new insights into the function of p53 in regulating stemness and animal development.
We previously generated Oct4-Venus Tg rats and established Oct4-Venus ESC lines in which Oct4 expression can be monitored by green fluorescence12,27. Here, both conventionally KO and mutant chimeras rats were generated using p53-null Oct4-Venus ESCs and their development was investigated. Moreover, an efficient method for a rapid generation of mutant chimeras was developed using ZFN-mediated gene targeting in rat ESCs. Using this method, developmental phenotypes can be observed within 1 month.
Results
Conventionally generated p53−/− females reveal the cause of NTDs
p53 homozygous KO rats were generated via germline transmission of heterozygous p53+/C ESCs (Fig. 1a,b,e). The details are described in the Materials and Methods. The Mendelian ratios in weaned rats produced from heterozygous intercrosses were investigated (Fig. 2a). The frequency of homozygous p53C/C rats was 16.9%, less than the anticipated value of 25% (Table 1). Moreover, only one p53C/C female developed normally, frequency = 0.70%, significantly less than 16.2% p53C/C males. These results suggest that most of the p53C/C females either do not survive gestation, or die after birth but prior to weaning. To investigate the developmental dysfunctions in p53C/C females, litters from heterozygous intercrosses were examined at embryonic day 16.0 (E16.0) to E18.0. Eleven p53C/C female embryos (12.8%, 11/86) were recovered at this stage; six (57%, 6/11) exhibited exencephaly (Table 1) and two of these also exhibited spina bifida (Fig. 2b). Although these two abnormalities are the most prevalent NTDs, spina bifida in p53 mutant mice has only been reported in one study28. Exencephaly was only found in the female embryos, consistent with previous observations of a higher incidence of NTDs in human females and in numerous mouse models29. Expression of SOX2, a marker for primordial neuronal cells expressed in the embryonic neural plate30, was detected on the surface of the brain and in areas of spina bifida (Fig. 2d, arrowheads), confirming that neural tube closure had failed. Compared to a p53+/C embryo (Fig. 2e, right), the aberrant ventricular zone (VZ) structure in the brain of a p53C/C exencephalic embryo was revealed by the localization of SOX2 (Fig. 2d left, arrows), which is expressed in the neuroendothelial stem cells of the VZ31. In this embryo, Oct4-Venus expressing cells were aberrantly located in the exencephalic region (Fig. 2b, green square and 2c).

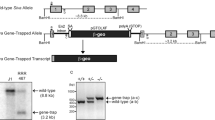
Schematic representation of p53 KO strategy in rats.
(a–d) Both mono- (b) and bi-allelic (c) or 2ndary (d) homologous recombination are induced by ZFN. (e, f) Heterozygous or homozygous ESC-injection leads to the generation of conventionally generated KO model (e) or ESC-based mutant chimeric models (f), respectively. A yellow box indicates a frame shift mutation induced by ZFNs. Pr., Primer. coKO, congenital KO. acKO, acquired KO.

Phenotypes in conventionally generated p53 homozygous rats.
(a) Schematic representation of heterozygous intercrosses indicates a loss of adult female. (b) An embryo at day 17.0 of gestation (E17.0) displaying exencephaly and spina bifida. A dotted green square indicates (c). (c) Fluorescence image of the area inside the dotted green square in (b). Oct4-Venus fluorescence is observed in the exencephalic region. (d) IHC staining for SOX2 identifies positive cells in the ventricular zone (arrows) and surface (arrowheads) of both brain and spina bifida (magnified image, scale bar = 100 μm).
Embryonic lethality in a mutant chimeric model
ZFNs can create site-specific double-strand breaks, which are repaired via non-homologous end joining, resulting in frame-shift mutations by the arbitrary addition or deletion of base pairs. Cotransfection of ZFNs with targeting vectors enhances homologous recombination, not only in human pluripotent cells32,33, but also in one-cell embryos, leading to the direct generation of knock-in mice34 and rats35. In the present work, ZFNs were used to produce homozygous mutant ESC lines by a single recombination step (Fig. 1c). Using this approach, 1 of 46 (2.2%) clones harbored dual knock-in alleles (p53C/C), while 7 of 46 (15%) clones possessed both knock-in and frame shift mutant alleles (p53C/Z). In a 2nd-step recombination, homozygous clones were also produced from a p53+/C3 ESC clone based on the same strategy using both ZFNs and a targeting vector expressing red fluorescence (Fig. 1b,d). A successful homologous recombination was achieved in 3 of 8 clones (38%, p53C/R; Supplementary Fig. S2c). These ESC lines were called acquired KO (acKO) ESCs (Fig. 1c). The contribution of p53−/− (p53C/C, p53C/Z, or p53C/R) ESCs to rats, which are called p53−/− (ESC) rats, was examined and the timeline for the rapid generation of the mutant chimeras is shown schematically (Fig. 3a,b). Microinjection of p53−/− ESCs into blastocysts led to the delivery of only a few pups (0.4 ± 0.2 per foster mother, n = 5, 4 cell lines). This number (0.4 ± 0.2/foster mother) was significantly smaller than the number of pups delivered following injection of p53+/+ ESCs into blastocysts (4.0 ± 1.1 per foster mother, n = 5, 3 cell lines, P<0.05) or p53+/− (p53+/C) ESCs (5.1 ± 1.1 per foster mother, n = 9, 4 cell lines, P<0.05) (Fig. 3c). The newborns derived from p53−/− ESC-injections did not exhibit a brown coat-color, indicating that they were not chimeras. Because these results suggest that the development of p53−/− (ESC) embryos was defective, fetal development at stages E14.0 to E17.0 was examined. Approximately 80% of the p53−/− (ESC) embryos (n = 14, 5 cell lines, Fig. 3d) showed abnormal development resulting in complete resorption (Fig. 3e, asterisks) or growth retardation (Fig. 3e, an arrow and inset). A large number of p53−/− ESC-derived cells were detected in these embryos (Fig. 3f, lower). However, the remaining p53−/− (ESC) embryos (30/189 embryos: 20.0 ± 5.6%) developed a normal body size (Fig. 3f, upper and Supplementary Table S1). The number of normal embryos (20.0 ± 5.6%) was significantly lower than that of p53+/+(ESC) embryos (53/63 embryos: 85.1 ± 5.8%, n = 4, 4 cell lines, P<0.01) or p53+/−(ESC) embryos (44/64 embryos: 69.0 ± 2.2%, n = 7, 4 cell lines, P<0.01) (Fig. 3d). Among the normal-sized p53−/−(ESC) embryos, 26 of 30 (87.8 ± 9.7%, n = 10, 5 cell lines) embryos were chimeras, whereas 22 of 26 displayed a relatively lower contribution of the mutant cells. Although the number of p53+/−(ESC) chimera (30/44 embryos, 66.9 ± 6.6%, n = 7, 4 cell lines) was similar to that of p53−/−(ESC) chimera (P = 0.14), the p53+/−(ESC) chimeras developed normally (Fig. 3c, d). The number of p53+/+(ESC) chimera (17/53 embryos, 36.0 ± 10.2%, n = 4, 4 cell lines) was significantly smaller than that of either p53+/−(ESC) (P = 0.044) or p53−/−(ESC) (P = 0.0081) chimera. These results suggest that p53 mutation enhances the chimeric contribution of ESCs and the high contribution of p53-null ESCs induces embryonic lethality.

Embryonic lethality in p53 mutant chimeras.
(a) Time line for generating p53−/−(ESC) chimeras and (b) Schematic representation to investigate phenotype during embryogenesis. (c) Number of neonates successfully delivered. p53+/+ (n = 5, 3 cell lines), p53+/− (n = 9, 4 cell lines) or p53−/− (n = 5, 4 cell lines) ESCs were injected into wild-type blastocysts. n, injection number. *, P <0.05, p53−/− vs. p53+/+ and p53+/−. (d) The ratio of chimeric embryos with normal body size at E14.0 to E17.0. p53+/+ (n = 4, 4 cell lines), p53+/− (n = 7, 4 cell lines) or p53−/− (n = 14, five cell lines) ESCs were injected into wild-type blastocysts. n, injection number. **, P <0.001, p53−/− vs. p53+/+ and p53+/−. (e) Developmental dysfunction in chimeric embryos injected with p53C/R2 ESCs at E16.0. An arrow indicates a chimera with growth retardation (inset). Asterisks indicate fetal absorption. (f) Correlation between developmental dysfunction and ESC contribution. p53C/C1 ESCs expressing AmCyan1 contribute to chimeric embryos at E14.0. An arrowhead indicates an ectopic expression of Oct4-Venus. (g) p53C/R4 ESC proliferation in blastocyst. Twelve ESCs were injected into blastocyst, followed by incubation overnight in YPAC medium. (h) Immunohistochemistry using Cleaved-Caspase3 antibody in liver of p53+/C(ESC) or p53C/C(ESC) chimera. Arrowheads indicate the apoptotic cells. (i) Spinal curvature (an arrow) in p53C/C(ESC) chimera. All scale bars = 100 μm.
To address the mechanisms by which p53−/− ESCs result in embryonic lethality, the behavior of p53−/− ESCs was followed in blastocysts incubated in vitro. Blastocysts were injected with 12 ESCs and incubated over night. Although p53+/+1 ESCs remained in the blastocysts, the number decreased to 4.0 ± 0.89 cells (Fig. 3g, upper, n = 5). In contrast, a significantly larger number of p53C/R4 ESCs (13.7 ± 1.2 cells, n = 7, P<0.0001) were detected (Fig. 3g, lower), indicating proliferation of the p53-null ESCs in the blastocysts. The excess proliferation may lead to a high ESC contribution, resulting in the developmental abnormalities that led to resorption of the fetuses. In fact, several of the p53C/C(ESC) chimeras with normal body size displayed increased number of apoptotic cells in the liver (Fig. 3h, arrowheads) and one chimera exhibited an abnormal spinal curvature (Fig. 3i, an arrow). Embryos such as these may die and undergo resorption before birth, resulting in the significant loss of neonates as shown in Figure 3d.
Morphology and global gene expression profile in p53−/− ESCs
The properties of p53−/− ESCs were examined. Venus-negative differentiated cells (Fig. 4a, left, arrowheads) survived the processes of cloning and passaging p53−/− Venus-positive ESCs (Fig. 4a, left, arrows), indicating that p53−/− differentiated cells escaped from apoptosis. Rat ESC colonies adopt a dome-shaped morphology and tend to detach from culture dishes coated with MEFs12,15. The p53−/− domed colonies were detached by pipetting and the cells were passaged after dissociation, leading to successful propagation of dome-shaped colonies; no differentiated cells were detected. (Fig. 4a, right, arrows). The morphology of the p53−/− lines was indistinguishable from that of p53+/− or p53+/+ cells (Supplementary Fig. S3). The p53C/C ESCs were positive for alkaline phosphatase (ALP) activity (Fig. 4b).

Characteristics of p53-null ESCs.
(a) A p53C/C1 ESC clone is shown. Arrows indicate pluripotent colonies. Arrowheads indicate differentiated cells. (b) ALP staining in p53C/C1 ESCs. (c) q-PCR analysis in p53 mutant ESCs. Transcript levels were normalized to Gapdh levels. Data are the mean ± SD of one biological sample assayed in four independent experiments. a, P<0.05 versus p53+/+; b, P<0.005 versus p53+/C. (d) Scatter plots of global gene expression microarrays comparing p53+/+ and p53C/C ESCs of an acKO (left) or coKO (right) line. The green lines delineate the boundaries of a 2-fold difference in gene expression levels. (e) Venn diagrams of the intersection between genes highly (left) or lowly (right) expressed in the coKO versus the acKO in p53C/C ESCs. (f) Developmental dysfunction in chimeric embryos injected with p53 C/C2 coKO ESCs at E16.0. All scale bars = 100 μm.
The expression levels of ESC marker genes, such as Oct4, Nanog and Sox2, were similar in p53C/C and p53C/R ESCs compared to p53+/+ or p53+/C cells, whereas loss of p53 mRNA and parallel reduction in the mRNA level of the p53 target gene p21 were confirmed in mutant ESC lines (Fig. 4c). The ESC line produced by acquired gene targeting in wild-type ESCs (‘acKO’ ESCs) and a congenital KO ESC line derived from heterozygous intercrosses (named coKO) were analyzed to determine whether some compensatory effect occurred in the coKO line. A microarray analysis showed that the coKO line had less divergent expression compared to the acKO line (acKO vs. coKO: 494 vs. 11 genes upregulated and 214 vs. 50 genes downregulated, Fig. 4e). Venn diagrams showing the overlap in genes identified in the two KO ESC datasets identified only five upregulated and 30 downregulated genes (Fig. 4e and see Supplementary Table S2). Many of the downregulated genes in the p53C/C ESCs were direct targets of p53, such as Puma, p21, Ccng1, Plk2, Phlda3 and Ptprv (Fig. 4d), whereas no genes for pluripotency or stemness were identified.
Chimera generation was used to investigate whether microinjection with these coKO ESCs could rescue mutant chimera development. Male ESC lines were also examined because homozygous males showed normal development. However, microinjection of both female cell lines (n = 4, 2 cell lines) and male coKO ESC cell lines (n = 7, 3 cell lines) produced chimeras in which embryogenesis failed, similar to the acKO chimeras (Fig. 4f and Supplementary Table S1). The fraction of full-sized embryos (15/68 embryos: 23.1 ± 4.0%, n = 9, 5 cell lines) was similar to that of acKO chimeras (30/189 embryos: 20.0 ± 5.6%, n = 14, 5 cell lines). These results indicate that lethality in mutant chimeras is due to abnormality of p53−/− ESCs.
p53−/− ESCs are resistant to apoptosis and differentiation
To investigate susceptibility to apoptosis, flow cytometry to detect surface exposure of Annexin V was performed in ESCs under routine culture conditions using YPAC medium [Y, Y-27632 (ROCK inhibitor); P, PD0325901 (MEK inhibitor); A, A-83-01 (TGF-β inhibitor); C, CHIR99021 (GSK3 inhibitor)]12. A control treatment with G418 caused an increase in Annexin V-positive apoptotic cells whereas each of the p53 genotype ESCs exhibited small population of the apoptotic cells (p53+/+, 11.0 ± 0.25%; p53+/C, 14.0 ± 0.30%; p53C/C, 11.0 ± 0.49%) (Fig. 5a). Assays for colony formation and embryoid body (EB) formation were performed under differentiation culture conditions to examine the behavior of the mutant ESCs. There was no genotype-dependent difference in the numbers of undifferentiated or differentiated colonies under conditions using YPAC medium and MEFs (Fig. 5b). Under culture conditions using Y media and MEFs (inhibitors P, A and C were absent, Fig. 5c), almost no p53+/+ undifferentiated colonies formed (1.7 ± 0.9 colonies) but some p53+/C colonies were observed (13.0 ± 2.1 colonies, P<0.01). Few differentiated colonies of either genotype were formed. In contrast, p53C/C cells formed a large number of both undifferentiated (56.0 ± 2.6 colonies, P<0.0001 vs. p53+/+; P<0.001 vs. p53+/C) and differentiated (26.3 ± 1.5 colonies, P<0.0001 vs. p53+/+; P<0.0001 vs. p53+/C) colonies (Fig. 5c). These results suggest that p53−/− ESCs strongly maintain both undifferentiated state and self-renewal capacities while the differentiated cells are protected from apoptosis, consistent with the results shown in Figure 3a. Next, colony formation was assessed under culture conditions in which ESCs weakly attach to un-coated culture dishes in the absence of MEFs. Although undifferentiated colony formation was rare in both p53+/+ (11.3 ± 3.5) and p53+/C (2.0 ± 1.2) ESCs, a large number of p53C/C ESCs formed colonies (96.7 ± 2.8 colonies, P<0.0001 vs. p53+/+; P<0.0001 vs. p53+/C; Fig. 4d). In the un-coated dishes in the absence of MEFs, differentiated colonies were rarely formed from any ESC genotype. Thus, the p53−/− ESCs might have an increased capacity to adhere tightly to the culture dish and/or proliferate without the support of feeder cells.

Colony and EB formation assays under differentiating conditions.
(a) Flow cytometry analysis. Annexin V-Cy5 was used to detect apoptotic cells (n = 3). (b–d) Colony formation assay. Five hundred cells were cultured under normal conditions (YPAC+MEF; b), differentiation conditions (Y+MEF; c), or apoptosis-inducing conditions (YPAC-MEF; d). A green or black bar indicates pluripotent or differentiated colony number, respectively (n = 3 or 4). (e, f) EB formation assay. EBs were formed from 2.5 × 105 cells (f, dotted line). Seven days after incubation without inhibitors, the cell number was counted (f, n = 3). Insets (e) indicate EBs at day 3. *, P <0.01. All scale bars = 100 μm.
When EB formation was examined, p53+/+ EBs formed by day 3 underwent apoptosis over time in culture, resulting in few EBs remaining by day 7 relative to day 3. In addition, Venus fluorescence was completely lost in these cells (Fig. 5e, left). In contrast, p53C/C EBs were large in size and number and maintained Venus fluorescence (Fig. 5e, right). The number of cells in p53C/C EBs (3.25 × 105) was significantly larger than cell number in p53+/+ EBs (1.00 × 103, P<0.01) or p53+/C EBs (2.47 × 104, P<0.01). Moreover, these data indicate that p53C/C EBs actively proliferated because the cell number increased from 2.5 × 105 at day 0 to 3.25 × 105 by day 7 (Fig. 5f). This result suggests that p53C/C cells are able to proliferate even in the absence of cell attachment. p53+/C EBs showed an intermediate phenotype with significant differences from the other genotypes (P<0.01 vs. p53+/+, P<0.01 vs. p53C/C).
Chromosomal instability in p53−/− cells
Next, karyotype analysis was performed in p53−/− cells. Although p53+/C ESCs maintained a normal karyotype 42,XX,[20], one p53C/R2 ESC clone exhibited abnormal karyotype 42,XX,add(15)(q22)[20] (Fig. 6a, red square and arrow). Moreover, once the p53C/R2 ESC clone differentiated under EB forming culture conditions for two weeks (Fig. 6b), an additional chromosomal aberration, 41,X,-X,add(15)(q22)[20], was found in all cells analyzed (Fig. 6a, blue square). In a p53C/C1 ESC clone, ESCs did not have an abnormal karyotype (42,XX[20]). However, cells derived from the p53C/C1(ESC) chimera in E14.0 rats displayed various chromosomal aberrations, such as 42,XX,add(1)(q52)[1], 42,XX,add(3)(p12) [1], 43,XX,+16[1] or 42,X,-X,+mar[1]. In cell cultures, p53+/+ cells derived from a recipient blastocyst were eliminated, resulting in occupation by p53C/C1 cells with AmCyan1 expression (Fig. 6c).

Chromosomal instability in p53-null cells.
(a) Cytogenetic analysis by G-band staining in p53C/R2 ESCs and EB-derived differentiated p53C/R2 cells. Abnormal chromosomes were indicated by red and blue squares. (b) Differentiated p53C/R2 EBs lacking Venus fluorescence. (c) Aberrant chromosomes were observed in cells derived from a p53C/C1(ESC) chimera at E14.0.
These findings demonstrate that p53−/− ESCs exhibit several features of abnormalities, such as blockage of differentiation, induction of chromosomal instability and escape from apoptosis, which are facilitated when the cells differentiate. Thus, p53 is indispensable for embryonic development in the mutant chimeric models (Fig. 1f) but dispensable in the homozygous models due to bypassing an ESC state (Fig. 1e).
Discussion
Here, two distinct strategies were used to generate p53 KO rats: conventionally generated homozygous KO and ESC-based mutant chimeras. In the homozyogous KO rats, NTDs such as exencephaly and spina bifida were observed. This is the first NTD model created in genetically modified rats. Previously, a 50% reduction in the number of females relative to males at weaning was observed in p53 homozygous KO mice17. In contrast, in the present study, a 96% reduction in the number of p53 homozygous KO female rats surviving to weaning relative to homozygous KO males was observed (Table 1). In rats, exencephaly occurred in a large fraction of the homozygous KO females (55%; Table 1), whereas only 8–16% of homozygous KO female mouse embryos exhibited exencephaly17,18. The survival ratio and spina bifida phenotype observed in these exencephalic rat embryos suggests that this species exhibits more severe phenotypes than mice. We hypothesize that rats are more sensitive to the stress of DNA damage than mice. Consistent with this observation, rat ESCs are more sensitive to differentiation signals than mouse ESCs, which is one reason why rat ESCs were not established until 2008. Mouse ESCs are very stable compared to other species. In mouse, successful chimera contribution and germline transmission using p53-null mouse iPSCs has been reported24,25. These results were unexpected, considering the vast amount of data regarding the effects of p53 on cell cycle arrest, apoptosis and DNA repair. In contrast, in the present study, mutant chimeras generated with rat ESCs demonstrated a clear phenotype of embryonic lethality, consistent with the data presented here showing the downregulation of p53 target genes, inhibition of apoptosis and differentiation and increase in chromosomal instability in p53-null rat ESCs or ESC-derived cells.
The rat is considered to be a better model than the mouse for many complex disorders that are common in humans36 and is currently the primary animal model in many preclinical tests, especially those related to cardiovascular disease, diabetes, breast cancer, chronic inflammatory diseases and age-related diseases20. Genetically modified rats are valuable platforms for the study of human physiology and disease. For example, in comparison to transgenic mice, transgenic rat models of Huntington disease not only present a more typical adult patient pathology but are also more suitable for in vivo metabolic and structural imaging20,37. In addition, Apc knockout mice develop tumors primarily in the small intestine, whereas both humans and rats develop colon cancer as a result of the Apc mutation38. These observations support the inconsistent phenotype of p53 mutant chimeras between rats and mice, as shown in the present work and suggest the importance of generating genetically modified rats to find novel gene functions.
In this study, the differences in the phenotypes of the p53 homozygous and mutant chimeric rat models were striking. Secondary mutations are accumulated in the mutant ESCs under in vitro culture conditions and in the differentiating cells during embryogenesis (Fig. 6). These aberrant cells are resistant to apoptosis due to p53 deficiency, which might lead to lethality of the mutant chimeras. These observations reflect the fact that a major p53 function is to be the “guardian of the genome”. Thus, the mutant chimeric strategy may prove useful in identifying authentic and/or novel gene functions. Finally, the present study demonstrated that mutant chimeric models can be generated within one month, circumventing both the risks associated with producing successful germline transmission as well as the time frame required for breeding both chimeras and heterozygous animals. In the mutant chimeric method, double or triple gene knockouts can be generated in a few months. These new combination strategies using embryonic stem cells, the mutant chimeric method and rats instead of mice will provide great insight into the novel functions of a large number of genes. The first example, shown here by deleting the p53 gene, provided new, substantial evidence demonstrating that p53 functions not only as the “guardian of the genome”, but also as the “guardian of the embryogenesis”.
Methods
Media, feeder cells, animals and primers
The YPAC medium was prepared by the addition of the following inhibitors: 10 μM Y-27632 (WAKO), 1 μM PD0325901 (Axon Medchem), 0.5 μM A-83-01 (TOCRIS) and 3 μM CHIR99021 (Axon Medchem) to a basic medium. The basic medium was composed of DMEM (including 110 mg/L sodium pyruvate and 200 mM GlutaMAX, GIBCO), 20% FBS (ES Cell Qualified Fetal Bovine Serum, Lot No. 1204059, GIBCO), 0.1 mM 2-mercaptoethanol (SIGMA), 1% nonessential amino acid stock (GIBCO) and 1× antibiotic antimycotic (GIBCO). Mitomycin C-treated MEFs resistant to neomycin (Millipore) were used as feeders and maintained in 10% FBS DMEM (Lot No. SFB30-1502, EQUITECH-BIO, INC.) with 1× antibiotic antimycotic. Animal experiments were performed in compliance with the guidelines of the Institute for Laboratory Animal Research, National Cancer Center Research Institute. These studies were approved by National Cancer Center Research Institute. All primer sequences are listed in Supplementary Table 3.
Generation of Oct4-Venus Tg rats and ESCs
Oct4-Venus Tg rats of the Wistar strain were generated via germline transmission of an Oct4-Venus ESC clone in the same manner described previously12. The Oct4-Venus ESC line derived from the Long-Evans Agouti (LEA) strain was generated in an earlier study12.
Establishment of rat ES cells from blastocysts
Rat blastocysts were gently flushed out from the uteri of E4.5 or E5.0 pregnant rats with a basic ES medium. After removal of the zona with acid Tyrode's solution (Ark Resource Co., Ltd.), whole blastocysts were plated onto 6-well plates and cultured on MEFs in basic ES medium with or without YPAC. After approximately 7 days, the blastocyst outgrowths were cut into pieces and replated under the same YPAC conditions. Emerging ESC colonies were then dissociated with Accutase (Innovative Cell Technologies, Inc.) and expanded. Domed and floated ESC colonies were detached from MEFs by pipetting, followed by routinely passaging every 3–4 days under MEF-YPAC conditions.
ZFN constructs and targeting plasmids
Custom-designed ZFN plasmids and ZFN-encoding mRNA for the rat p53 gene were purchased from Sigma-Aldrich. The design, cloning and validation of the ZFNs were performed by Sigma-Aldrich. The ZFN pair recognizing exon 4 of the p53 gene was 5′: TTCTCCAGTCTTCCTCCAG, 3′: ATTCTGGTAAGGAGCCGG. The targeting donor was composed of the CAG-AmCyan1-IRES-Neo-pA or CAG-tdTomato-IRES-Neo-pA cassette with short homology 5′ (736 bp) and 3′ (711 bp) arms. These homology arms were amplified from rat genomic DNA using the KOD Ver.2 DNA polymerase PCR system (Toyobo). The sequences of these primers are listed in Supplementary Table S3. Both arms were set at several base pairs from a ZFN-induced cleavage site.
Introduction of the targeting vector and ZFNs into LEA rat ESCs and generation of p53 heterozygous rats
To disrupt exon 4 of the p53 gene, 5 μg of ZFN-encoding mRNAs and 10 μg of a targeting plasmid (5′arm-CAG-AmCyan1-IRES-Neo-pA-3′arm cassette) were co-transfected into 6.5 × 105 of Oct4-Venus ESCs derived from a LEA female strain at passage 5 with a Mouse ESC Nucleofector Kit (Amaxa Inc.). The cells were plated on MEFs in YPAC medium with 2% Matrigel (BD Biosciences) to keep the ESC colonies adhered to the MEFs. One day after nucleofection, geneticin was added to the YPAC medium at 0.2 μg/ml. Eleven days after nucleofection, geneticin-resistant colonies were selected using handmade capillaries and expanded. Picking and expanding seven colonies provided four (57%) heterozygous (p53+/C) clones and two (29%) homozygous clones harboring both knock-in and frame shift mutation alleles (p53C/Z) (Supplementary Fig. S1a). The ZFN-induced frame shift mutation was identified by band shift in PCR analysis (Supplementary Fig. S1a, No. 6 clone, asterisk) and sequence analysis (Supplementary Fig. S1c) or using a SURVEYOR Mutation Detection Kit (Supplementary Fig. S1b, No. 8 clone). A p53+/C ESC clone was used for microinjection, resulting in the generation of p53+/C rats through chimeric germline transmission.
Introduction of the targeting vector and ZFNs into Wistar rat ESCs
For nucleofection, 10 μg of a targeting plasmid (5′arm-CAG-AmCyan1-IRES-Neo-pA-3′arm cassette) and 5 μg of ZFN-encoding mRNAs were co-transfected into 4.5 × 106 of Oct4-Venus ESCs (Wistar) at passage 3. As a control experiment, 10 μg of the targeting plasmid without ZFNs was transfected into 4.5 × 106 of Oct4-Venus ESCs at passage 3. One out of 46 (2.2%) clones was p53C/C, while seven of 46 (15%) clones were p53C/Z. A sequence data revealed an 8-bp deletion in the p53C/Z1 clone (Figure S2B). This ZFN-induced small deletion was also confirmed by a downward band shift (indicated by asterisks in Supplementary Fig. S2a). As a control experiment, the targeting vector alone was introduced without ZFN. Although 14 geneticin-resistant colonies appeared, they did not achieve homologous recombination (Supplementary Fig. S2a, lane 5). Knockout clones were also produced using a 2nd-step recombination by introducing the 10 μg of targeting plasmid (5′arm-CAG-tdTomato-IRES-Neo-pA-3′arm cassette) and 5 μg of ZFN-encoding mRNAs into 2.5 × 106 cells of a p53+/C3 ESC clone at passage 9 (Fig. 6). Eight red fluorescence (tdTomato)-positive clones were chosen without geneticin selection and successful homologous recombination was achieved in three of these (38%, p53C/R; Supplementary Fig. S2c).
Surveyor nuclease (Cel-I) assay
A ZFN target locus was amplified by PCR (35 cycles: 10 s denaturing at 98°C, 30 s annealing at 62°C and 1 min elongation at 72°C) using primers 1 and 5 (Fig. 6 and Supplementary Table S3). The Cel-I assay was carried out following the manufacturer's protocol (TRANSGENOMIC, Inc.).
ALP staining, immunohistochemistry (IHC) and Annexin V-apoptosis assay
Cells were fixed in 4% paraformaldehyde. ALP staining was performed with the Vector Blue substrate (Vector Labs) according to the manufacturer's instructions. Formalin-fixed and paraffin-embedded slides were stained with hematoxylin and eosin or used for IHC. Antigen retrieval was performed by autoclave in a sodium citrate buffer. The slides were incubated with Sox2 (BioLegend, 1:200) or Cleaved Caspase-3 (Cell Signaling, 1/1000) primary antibody at 4°C overnight. The next day, after washing, the samples were incubated with horseradish peroxidase-conjugated secondary antibody for 1 h. They were then washed and incubated with 3,3′-diaminobenzidine tetrahydrochloride DAB (Thermo Scientific). An assay for apoptotic ESCs was performed using Annexin V-Cy5 following the manufacturer's protocol (BioVision). Pluripotent ESC colonies were solely harvested and dissociated with Accutase, followed by incubating 1–5 × 105 cells with the Annexin V-Cy5 for 5 min in the dark.
Q-PCR analysis
Total RNA was isolated using ISOGEN (Nippongene). cDNA was synthesized with 2 μg of the total RNA using Super Script III RT (Invitrogen) and oligo-dT primer (Invitrogen). cDNAs were used for PCR using Platinum SYBR Green qPCR SuperMix UDG (Invitrogen). Optimization of the q-PCR reaction was performed according to the manufacturer's instructions (PE Applied Biosystems, Tokyo, Japan). All quantitations were normalized to an endogenous control GAPDH.
Microarray analysis
A one-color microarray-based gene expression analysis system (Agilent Technologies) using SurePrint G3 Rat GE 8 × 60 K Kit containing 30507 probes (26930 genes) was used following the manufacturer's instructions.
EB formation
After ES cells were dissociated into single cells using Accutase, 5 × 105 cells were cultured in PAC medium. After overnight incubation, the EB contained media were separated and cultured in media with or without PAC on a low cell-binding dish (NUNC). After 7 days of incubation, the cell number of EBs was counted after dissociation with Accutase.
Chimera production
In all blastocyst injection experiments, 12 ESCs were injected into E4.5 blastocysts. YPAC or PAC inhibitors were constantly included in media during both microinjection and blastocyst incubation. ESC-injected blastocysts were transferred to E3.5 pseudo-pregnant rats. The contribution of ESCs to the resulting chimeras was determined by the appearance of coat-color or fluorescence.
Karyotype analyses in p53−/− cells
G-band staining was performed in cultured cells from embryos, ESCs, or ESC-derived differentiated cells. Head of E14.0 chimeric embryo was dissociated with Accutase and karyotype analysis was examined in the cells at passage 4. p53C/R2 ESCs at five passages after the generation of the gene-targeted null mutation or EB-derived differentiated p53C/R2 cells at seven passages were analyzed. EBs were formed at passage 5 and cultured for 2 weeks, followed by two passages to expand the cells. The differentiated state was confirmed by a loss of Oct4-Venus expression, as well as by the cell morphology.
Statistical analysis
Results are given as the mean ± SD. Statistical analysis was conducted using Student t-tests. P < 0.05 was considered significant.
References
Cozzi, J. et al. Pronuclear DNA injection for the production of transgenic rats. Methods Mol. Biol. 561, 73–88 (2009).
Dann, C. T., Alvarado, A. L., Hammer, R. E. & Garbers, D. L. Heritable and stable gene knockdown in rats. Proc. Natl. Acad. Sci. U S A. 103, 11246–11251 (2006).
Zan, Y. et al. Production of knockout rats using ENU mutagenesis and a yeast-based screening assay. Nat. Biotechnol. 21, 645–651 (2003).
van Boxtel, R., Gould, M. N., Cuppen, E. & Smits, B. M. ENU mutagenesis to generate genetically modified rat models. Methods Mol. Biol. 597, 151–167 (2010).
Kitada, K. et al. Transposon-tagged mutagenesis in the rat. Nat. Methods 4, 131–133 (2007).
Kitada, K., Keng, V. W., Takeda, J. & Horie, K. Generating mutant rats using the Sleeping Beauty transposon system. Methods 49, 236–242 (2009).
Izsvák, Z. et al. Generating knockout rats by transposon mutagenesis in spermatogonial stem cells. Nat. Methods 7, 443–445 (2010).
Geurts, A. M. et al. Knockout rats via embryo microinjection of zinc-finger nucleases. Science 325, 433 (2009).
Mashimo, T. et al. Generation of knockout rats with X-linked severe combined immunodeficiency (X-SCID) using zinc-finger nucleases. PLoS One 5, e8870 (2010).
Buehr, M. et al. Capture of authentic embryonic stem cells from rat blastocysts. Cell 135, 1287–1298 (2008).
Li, P. et al. Germline competent embryonic stem cells derived from rat blastocysts. Cell 135, 1299–1310 (2008).
Kawamata, M. & Ochiya, T. Generation of genetically modified rats from embryonic stem cells. Proc. Natl. Acad. Sci. USA 107, 14223–14228 (2010).
Hamanaka, S. et al. Generation of germline-competent rat induced pluripotent stem cells. PLoS One 6, e22008 (2011).
Hirabayashi, M. et al. Rat transgenesis via embryonic stem cells electroporated with the Kusabira-orange gene. Mol. Reprod. Dev. 77, 474 (2010).
Tong, C., Li, P., Wu, N. L., Yan, Y. & Ying, Q. L. Production of p53 gene knockout rats by homologous recombination in embryonic stem cells. Nature 467, 211–213 (2010).
Donehower, L. A. et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356, 215–221 (1992).
Armstrong, J. F., Kaufman, M. H., Harrison, D. J. & Clarke, A. R. High-frequency developmental abnormalities in p53-deficient mice. Curr. Biol. 5, 931–936 (1995).
Sah, V. P. et al. A subset of p53-deficient embryos exhibit exencephaly. Nat. Genet. 10, 175–180 (1995).
Donehower, L. A. & Lozano, G. 20 years studying p53 functions in genetically engineered mice. Nat. Rev. Cancer 9, 831–841 (2009).
Huang, G. et al. Beyond knockout rats: new insights into finer genome manipulation in rats. Cell Cycle 10, 1059–1066 (2011).
van Boxtel, R. et al. Homozygous and heterozygous p53 knockout rats develop metastasizing sarcomas with high frequency. Am. J. Pathol. 179, 1616–1622 (2011).
Puzio-Kuter, A. M. & Levine, A. J. Stem cell biology meets p53. Nat. Biotechnol. 27, 914–915 (2009).
Lin, T. et al. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat. Cell Biol. 7, 165–171 (2005).
Hong et al. Suppression of induced pluripotent stem cell generation by the p53–p21 pathway. Nature 460, 1132–1135 (2009).
Marion et al. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature 460, 1149–1153 (2009).
Kawamata, M. & Ochiya, T. Establishment of embryonic stem cells from rat blastocysts. Methods Mol. Biol. 597, 169–177 (2010).
Kawamata, M. & Ochiya, T. Gene-manipulated embryonic stem cells for rat transgenesis. Cell Mol. Life Sci. 68, 1911–1915 (2011).
Hosako, H. et al. The roles of p53 and p21 in normal development and hyperthermia-induced malformations. Birth Defects Res. B Dev. Reprod. Toxicol. 86, 40–47 (2009).
Chen, X. et al. Sex difference in neural tube defects in p53-null mice is caused by differences in the complement of X not Y genes. Dev. Neurobiol. 68, 265–273 (2008).
Takemoto, T. et al. Tbx6-dependent Sox2 regulation determines neural or mesodermal fate in axial stem cells. Nature 470, 394–398 (2011).
Miyagi, S. et al. The Sox2 regulatory region 2 functions as a neural stem cell-specific enhancer in the telencephalon. J. Biol. Chem. 281, 13374–13381 (2006).
Hockemeyer, D. et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat. Biotechnol. 27, 851–857 (2009).
Soldner, F. et al. Generation of Isogenic Pluripotent Stem Cells Differing Exclusively at Two Early Onset Parkinson Point Mutations. Cell 146, 318–331 (2011).
Meyer, M., de Angelis, M. H., Wurst, W. & Kühn, R. Gene targeting by homologous recombination in mouse zygotes mediated by zinc-finger nucleases. Proc. Natl. Acad. Sci. USA 107, 15022–15026 (2010).
Cui, X. et al. Targeted integration in rat and mouse embryos with zinc-finger nucleases. Nat. Biotechnol. 29, 64–67 (2011).
Abbott, A. Return to the rat. Nature 460, 788 (2009).
von Horsten, S. et al. Transgenic rat model of Huntington's disease. Hum. Mol. Genet. 12, 617–24 (2003).
Amos-Landgraf, J. M. et al. A target-selected Apc-mutant rat kindred enhances the modeling of familial human colon cancer. Proc. Natl. Acad. Sci. U.S.A. 104, 4036–41 (2007).
Acknowledgements
We thank F. Takeshita, T. Katsuda, K. Hagiwara, Y. Yoshioka, R. Takahashi, N. Kosaka, H. Tsuda, H. Sasaki and Y. Tamai for their technical advice. This work is supported by a Grant-in-Aid from the Third-Term Comprehensive 10-Year Strategy for Cancer Control.
Author information
Authors and Affiliations
Contributions
M.K. designed and performed experiments. M.K. and T.O. wrote the manuscript. T.O. supervised the project.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplemental info TableS1-3 and FigureS1-3
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Kawamata, M., Ochiya, T. Two distinct knockout approaches highlight a critical role for p53 in rat development. Sci Rep 2, 945 (2012). https://doi.org/10.1038/srep00945
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep00945
This article is cited by
-
Generation of Hprt-disrupted rat through mouse←rat ES chimeras
Scientific Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
