
Abstract
Recent data have shown that TLR4 performs a key role in cerebral ischemia-reperfusion injury which serves as the origin of the immunological inflammatory reactions. However, the therapeutic effects of pharmacological inhibitions of TLR4 and its immediate down-stream pathway remain to be uncovered. In the present study, on mice, intracerebroventricular injection of resatorvid (TLR4 signal inhibitor; 0.01 μg) significantly reduced infarct volume and improved neurological score after middle cerebral artery occlusion and reperfusion. The levels of phospho-p38, nuclear factor-kappa B and matrix metalloproteinase 9 expressions were significantly suppressed in the resatorvid-treated group. In addition, NOX4 associates with TLR4 after cerebral ischemia-reperfusion seen in mice and human. Genetic and pharmacological inhibitions of TLR4 each reduced NOX4 expression, leading to suppression of oxidative/nitrative stress and of neuronal apoptosis. These data suggest that resatorvid has potential as a therapeutic agent for stroke since it inhibits TLR4-NOX4 signaling which may be the predominant causal pathway.
Similar content being viewed by others


Introduction
Stroke, including cerebral infarction, is the one of the commonest causes of death in developed countries. However, there is no curative therapeutic strategy for patients who have suffered cerebral ischemia, other than thrombolysis using recombinant tissue plasminogen activator (rtPA). Therefore, novel therapeutic options for ischemic stroke are globally required.
Ischemia-reperfusion injury provokes inflammatory cascades and consequent tissue damage. These reactions are initiated by endogenous ligands known as damage-associated molecular patterns, such as high-mobility group box 1 (HMGB1) and heat shock proteins, which are released from necrotic cells and then activate the innate immune system via pattern recognition receptors1,2. We and others have demonstrated in gene ablation mice that TLR4, by triggering transduction cascades and inflammatory cytokines, performs a pivotal role in the pathogenesis of focal cerebral ischemia3,4,5,6,7. In addition, it has been reported that the brain tissue damage induced by HMGB1 is abolished by a deletion of TLR4 function, suggesting that among the innate immune receptors, TLR4 plays a central role in the deterioration that follows ischemia and reperfusion8,9. Moreover, TLR4 is expressed not only on immune cells, but also on non-immune cells, especially neurons, indicating that activation of TLR4, both directly and indirectly, leads to neuronal cell death4,7,8. Although many reports have provided strong evidence that TLR4 plays an essential role in the progression of such ischemic injury, no study has yet explored pharmacological approaches targeting TLR4 signaling at a potentially therapeutic time during focal cerebral ischemia.
The pathophysiological process of ischemia followed by reperfusion generates reactive oxygen species (ROS), which are chemically reactive molecules including superoxide anion (O2-), hydrogen peroxide (H2O2) and hydroxyl radical (HO)10,11. Reactive nitrogen species (RNS) are also produced by the reaction of nitric oxide (NO) with the superoxide anion (O2-) to form peroxynitrite (ONOO-)12. The robust production of ROS and RNS which cause oxidation and nitration of DNAs, lipids and proteins; events often referred to as oxidative/nitrative stress is a key contributor to the critical damage to brain tissue and neurological functions that can occur in focal ischemia10,11,12. However, despite being effective scavenging agents in the laboratory, almost all attempts at their clinical development have been unsuccessful13.
In recent years, another approach targeting oxidative/nitrative stress has been tried in ischemic stroke: inhibiting the formation of ROS/RNS14,15. The primary form of the free radical is superoxide anion (O2−), which is mainly formed by the action of NADPH oxidase (NOX)16. Six homologs (NOX1, NOX3, NOX4, NOX5, DUOX1 and DUOX2) of gp91phox (NOX2) have been identified in various non-phagocytic cells16. NOX4 expression is increased after cerebral ischemia and NOX4-deficient mice exhibit significantly less serious ischemic injury than their controls, indicating that preventing the generation of ROS/RNS early in the process by blocking NOX4 is a potential therapeutic strategy15. In addition, NOX4 has been shown to be required, via its interaction with TLR4, for both endogenous and exogenous TLR4 ligand-induced ROS generation17,18. On the basis of these findings, we hypothesized that TLR4 mediates the pathway to injury and that interaction between TLR4 and NOX4 allows the endogenous ligands released in cerebral ischemic conditions to activate a pathway leading to the production of ROS/RNS.
Resatorvid, a cyclohexene derivative (Fig. 1A), is a TLR4 signal inhibitor originally developed to halt the progression of severe sepsis19. We began the present study by synthesizing this compound. Then, we used a mouse model of middle cerebral artery occlusion (MCAO) (a) to assess whether resatorvid might reduce infarct volume and/or neurological deficits and if so, (b) to elucidate the underlying mechanisms including transduction factors and involvement of matrix metalloproteinase 9 (MMP9). We also investigated the association of TLR4 with NOX4 to clarify the putative pathway immediately down-stream of TLR4 that is activated in cerebral ischemia, using from the mouse model and from human patients tissue.

Effects of resatorvid, a TLR4 signal inhibitor, on infarction at 22 h after ischemia and reperfusion in mice.
(A) Chemical structure of resatorvid {ethyl (6R)-6-[N-(2-chloro-4-fluorophenyl)sulfamoyl]cyclohex-1-ene-1-carboxylate}. (B) TTC staining of coronal brain sections (2 mm thick) at 22 h after ischemia and reperfusion in representative mice. Upper panels, vehicle-treated. Lower panels, resatorvid-treated (at 0.01μg, i.c.v.). (C) Brain infarct area measured at 24 h after MCAO. Brains were removed and the forebrains sliced into five coronal 2-mm sections. Sections are identified according to the distance from the frontal limit of the forebrain. (D, E) Resatorvid-treated group (at 0.01 ug, i.c.v.) exhibited decreased infarct volume and improvement in neurological deficits vs. vehicle. * P < 0.05, ** P< 0.01 vs. vehicle, n = 9 to 11. Statistical significance was determined by a one-way ANOVA with Dunnett’s test or the Mann-Whitney U-test.
Results
Treatment with the TLR4 inhibitor resatorvid attenuated cerebral ischemic damage
Mice were subjected to 2 h of ischemia followed by 22 h of reperfusion. To determine the effect of inhibiting TLR4 signaling after the ischemia, we injected one of several doses of resatorvid, a TLR4 signal inhibitor, intracerebroventricularly (i.c.v.) into mice just after reperfusion. The TTC staining and neurological score results showed no clear differences between the resatorvid-treatment (0.001 or 0.003 ug, i.c.v) and vehicle-treatment groups (Fig. 1C to E). However, at 0.01 ug, i.c.v., resatorvid decreased the cerebral infarction, by approximately 40%, at 24 h after cerebral ischemia (Fig. 1B to D). Moreover, at 0.01 ug, i.c.v., but not at 0.001 or 0.003 ug, i.c.v., resatorvid improved the neurological deficits (Fig. 1E). Based on infarction measurements, resatorvid significantly reduced both the infarct area and volume in a dose-related manner, with significant effects being seen at 0.01 ug (Fig. 1C to D). There was no significant difference in blood pH, pCO2, pO2, or rCBF, or systemic blood pressure, between the vehicle-treated group and the resatorvid-treated group.
Pharmacological inhibition of TLR4 activation by resatorvid after cerebral ischemia reduced the ensuing signaling
We and others have recently reported that TLR4 activates nuclear factor-kappa B (NF-κB) and thereby contributes to ischemic injury4,7. To estimate the mechanism responsible for the above effects of resatorvid, we evaluated the level of the p65 subunit nuclear factor-κB (NF-κB) in nuclear and cytoplasmic fractions by Western blotting. In the vehicle group, the p65 expression level in the nuclear fraction was significantly greater in ipsilateral brain tissue than in tissue from the non-ischemic contralateral side (Fig. 2A). At 22 h after ischemia and reperfusion, resatorvid at 0.01 ug, i.c.v. significantly reduced the level of p65 in the nuclear fraction compared with that in the vehicle group (Fig. 2A). We also investigated the expression level of IRF-3, but no difference was found between the ipsilateral and contralateral sides (Fig. 2B). Resatorvid did not affect either of those expression levels on the contralateral side (Fig. 2A to B). It has been reported that MAPKs and their transcriptional factors, such as p38 and c-jun, are phosphorylated and activated in the ischemic region and that the phosphorylated proteins may play key roles in the evolution of brain damage following cerebral ischemia20. We measured the phosphorylation levels of p38 and c-jun in the ischemic hemisphere. Our quantitative analysis revealed significantly upregulated levels of p-p38 and p-c-jun at 22 h after ischemia and reperfusion (Fig. 2C to D). Resatorvid treatment at 0.01 ug, i.c.v. significantly prevented the ischemia and reperfusion-induced p38 phosphorylation and tended to reduce c-jun phosphorylation (Fig. 2C to D). It is known that activation of MMP9 has detrimental effects on stroke outcome21,22. It has recently been reported that upon cerebral ischemia, MMP9 may be directly induced by HMGB1, which is an endogenous immune ligand released by necrotic cells, mainly via TLR48. We investigated whether resatorvid might reduce MMP9 activity after cerebral ischemia. Resatorvid at 0.01 ug, i.c.v. significantly decreased the level of MMP9 expression seen after ischemia and reperfusion compared with that seen in the vehicle-treated group (Fig. 2E).

Pharmacological inhibition by resatorvid of TLR4 activation reduced down-stream signaling after cerebral ischemia.
(A) Western blots of nuclear (Nu) and cytoplasmic (Cyto) fractions show increased expression levels of the p65 subunit of NF-κB were on the ipsilateral side (Ipsi) in vehicle-treated mice [vs. the contralateral side (Contra)]. Quantitative analysis showed that p65 expression level in the nuclear fraction on the ipsilateral side was lower in the resatorvid-treated group (at 0.01 μg, i.c.v.) than in the vehicle group. (B) There was no clear difference in the level of IRF-3 expression between the ischemic and non-ischemic sides in the vehicle group. (A, B) Resatorvid at 0.01 μg had no effect on the contralateral side. # and *, P< 0.05 (as shown), n = 7. (C) The p38 phosphorylation level was higher in the vehicle-treated group than in the sham-operated group. Resatorvid (at 0.01 μg, i.c.v.) significantly decreased it, compared to vehicle treatment. (D) Treatment with resatorvid (at 0.01 μg, i.c.v.) tended non-significantly to reduce the c-jun phosphorylation level, which had been increased by cerebral ischemia. (E) Ischemia plus reperfusion increased MMP9 expression and resatorvid (at 0.01 μg, i.c.v.) down-regulated the MMP9 expression at 22 h after reperfusion. # P< 0.05, ## P < 0.01 vs. sham, * P< 0.05, ** P < 0.01 vs. vehicle, n = 7 to 12. Statistical significance was evaluated with Student unpaired, two-tailed t-test.
Association of TLR4 with NOX4 in cerebral ischemia
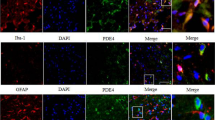
NADPH oxidases (NOX) are a major enzymatic source of reactive oxygen species (ROS) and NOX4 which is one of the 7 isoforms of the NADPH (nicotinamide adenine dinucleotide phosphate) oxidase family but not NOX1 or NOX2, may play a central role in cerebral ischemia15. Recent evidence has shown that TLR4 interacts with NOX4 not only upon lipopolysaccharide (LPS) stimulation but also in renal ischemia, leading in each case to oxidative stress17,18. Those reports led us to explore the possible association between TLR4 and NOX4 in cerebral ischemia. By double-immunostaining, TLR4 was found to be co-localized with NOX4 in the cortex of ischemic mice (Fig. 3A). In contrast, TLR4- or NOX4-positive cells were not detected in the cortex of sham-control mice (Fig. 3A). Notably, the same co-localization was detected in ischemic cortex from acute human stroke patient, but not in non-ischemic cortex from human control patient (Fig. 3A). Immunoprecipitation showed that TLR4 interacts with NOX4 in ischemic brain tissue (Fig. 3B). It has recently been reported that TLR4 activation may affect the level of NOX4 expression18. Here, we investigated whether TLR4 might induce NOX4 expression in cerebral ischemia in mice. In wild-type mice, NOX4 was significantly increased at 24 h after cerebral ischemia (Fig. 3C). However, in TLR4 KO mice, there was no clear difference between the ischemic group and the sham-operated group. The expression level of NOX4 after ischemia and reperfusion was decreased in TLR4 KO mice (Fig. 3C). The increased level of NOX4 expression induced by cerebral injury was significantly suppressed in the resatorvid-treated group (at 0.01 μg, i.c.v.) versus the vehicle-treated group (Fig. 3D).

Association of TLR4 with NOX4 in cerebral ischemia.
(A) Representative photographs showing double-immunostaining of TLR4 and NOX4 in sham-operated mice, ischemic mice, human control and human stroke patient. NOX4 was increased and co-located with TLR4 in the murine ischemic model and in the human stroke patient. Scale bar = 10 μm. (B) Brain tissues from a sham-operated or ischemic mouse hemisphere were subjected to immunoprecipitation using an antibody against TLR4. The immunoprecipitation material was then subjected to western blot analysis and proteins were detected using an anti-NOX4 antibody. TLR4 interacted with NOX4 in brain tissue after ischemia. (C) Comparison of TLR4 KO mice with wild-type mice as regards NOX4 expression after cerebral ischemia and reperfusion. # P< 0.05, vs. sham, n = 5. (D) Effects of resatorvid on NOX4 expression at 24 h after MCAO. Treatment with resatorvid (at 0.01 μg, i.c.v.) suppressed the NOX4 upregulation seen after cerebral ischemia plus reperfusion. ## P < 0.05, ## P < 0.01 vs. sham, * P< 0.05, vs. ischemia/reperfusion control or vehicle, n = 10. Statistical significance was evaluated with Student unpaired, two-tailed t-test.
TLR4 inhibition reduced cerebral ischemia-induced oxidative/nitrative stress
The overproduction of reactive oxygen species (ROS) that results from cerebral ischemia induces oxidative/nitrative stress and this damages DNAs, proteins and lipids, leading to neuronal degeneration10,11. Because TLR4 inhibition downregulated the expression of NOX4, one of the oxidases that lead to ROS generation, we next evaluated oxidative/nitrative stress after cerebral ischemia. Immunohistochemical analysis of the cortical peri-infarct region of mice revealed that TLR4 deficiency reduced the number of cells positive for 8-OHdG/nitrotyrosine, which are markers of oxidative/nitrative stress (Fig. 4A to B). Resatorvid at 0.01 ug, i.c.v. reduced that number (Fig. 4C to D). Taken together, these findings indicate that TLR4 controls oxidative/nitrative stress via NOX4 and that both genetic and pharmacological inhibitions of TLR4 suppresses such stress.

Genetic and pharmacological inhibition of TLR4 signaling led to a reduction of oxidative/nitrative stress after ischemia plus reperfusion.
(A to D) Representative photographs show 8-OHdG-positive or nitrotyrosine-positive cells in the cortical peri-infarct region of mice. (A, C) 8-OHdG-positive cells were increased by ischemia plus reperfusion. (B, D) Nitrotyrosine-positive cells were increased by ischemia plus reperfusion. (A, B) TLR4 KO mice exhibited significantly reduced (A) oxidative and (B) nitrative stress compared to wild-type mice. (C, D) Pharmacological inhibition of TLR4 by resatorvid (at 0.01 μg, i.c.v.) reduced (C) oxidative and (D) nitrative stress compared to vehicle treatment. Scale bar = 20 μm. ## P < 0.01 vs. sham, ** P < 0.01 vs. wild-type (A, B), vs. vehicle (C, D), n = 5. Statistical significance was evaluated with Student unpaired, two-tailed t-test.
Resatorvid prevented neuronal apoptosis
The oxidative/nitrative stress induced by overproduction of both ROS and RNS results in neuronal apoptosis in cerebral ischemia10,11. We examined the expression of cleaved caspase-3, an apoptosis marker and its co-location with NeuN, a neuronal marker, to evaluate the number of apoptotic neuronal cells in the peri-infarct zone of the cortex. The resatorvid-treated group (at 0.01 μg, i.c.v.) exhibited significantly decreased neuronal apoptosis after ischemia and reperfusion versus the vehicle group (Fig. 5A to B).

Resatorvid reduced neuronal cell death after cerebral ischemia.
(A) Representative photographs showing immunolabels for the apoptosis marker cleaved caspase-3, the neuronal marker NeuN and the nuclear marker Hoechst33342 in the cortical peri-infarct region of sham-operated, vehicle-treated and resatorvid-treated mice. (B) Quantitative analysis of neuronal apoptosis at 24 h after MCAO. Cleaved caspase-3-positive neuronal cells were increased by 2 h of ischemia plus 22 h of reperfusion. Treatment of mice with resatorvid (at 0.01 μg, i.c.v.) partly prevented the neuronal apoptosis. Scale bar = 20 μm. ## P < 0.01 vs. sham, ** P < 0.01 vs. vehicle, n = 5. Statistical significance was evaluated with Student unpaired, two-tailed t-test.
Discussion
In the present study, we revealed that resatorvid, a TLR4 inhibitor, significantly reduced the neuronal damage occurring after cerebral ischemia, a form of stroke. This is the first study to evaluate the effects of pharmacological TLR4 inhibition on the neuronal damage induced by cerebral ischemia and reperfusion. Accumulating evidence suggests that TLR4, an essential component of innate immunity, may play a central role in cerebral ischemia3,4,5,6. Similarly, we recently reported that TLR4 KO, but not TLR3 KO or TLR9 KO, mice exhibit neuroprotection against cerebral ischemia7. Figure 6 summarizes the putative mechanisms involving TLR4 that might be brought into play after cerebral ischemia, based on data from previous studies and the present one. Our finding that inhibition of TLR4 signaling after ischemia and reperfusion was protective against the neuronal damage induced by focal cerebral ischemia suggests that activation of TLR4 signaling occurring after reperfusion contributes to the pathogenic deterioration associated with cerebral ischemia and that such signaling is therefore a potential therapeutic targets of the stroke. Furthermore, we demonstrated that NOX4 may have a key role for TLR4-mediated inflammation in cerebral ischemia.

Putative mechanism for TLR4 signaling and effect of resatorvid on transient focal ischemia.
TLR4 is activated in response to damage-associated molecular patterns (DAMPs). NOX4 is induced by and interacts with, TLR4 in cerebral ischemia. TLR4-NOX4 pathway leads to inflammatory cascades, resulting in brain damage. Resatorvid binds to intracellular domain of TLR4 and interferes with the ensuing cascades, thereby protecting against ischemic damage.
The TLR4 signaling induced by endogenous ligands reportedly activates mitogen-activated protein kinases (MAPKs) and their transcription factors in cerebral ischemia5,7. Further, it has been reported that MMP9 is upregulated by HMGB1, an endogenous ligand, through TLR4 activation8. Although it is difficult to differentiate between changes that are causing the smaller infarct versus changes caused by the smaller infarct, the present data suggest that the pharmacological inhibition of TLR4 induced by resatorvid significantly suppressed the up-regulation of NF-κB in the nuclear fraction, the phosphorylation of p38 and the increased MMP9 expression seen after ischemia and reperfusion, with the result that resatorvid partly prevented the neuronal apoptosis induced by the cerebral ischemia. We have provided insights into the putative mechanisms underlying the neuroprotection (Figure 6).
Previously, resatorvid has been found to protect against the effects induced by lipopolysaccharide (LPS), an exogenous TLR4 ligand, in a systemic inflammation model19. Resatorvid blocks TLR4 signaling by binding directly to a specific amino acid, Cys747, in the intracellular domain of TLR4 and thereby disrupts the interaction of TLR4 with adaptor molecules, leading to suppression of transduction factors and the down-stream pro-inflammatory mediators, such as nitric oxide and multiple cytokines23.
However, the signaling pathway immediately down-stream of TLR4 in transient focal ischemia has remained unclear despite many studies approaching an answer. It is known that pathways involving myeloid differentiation factor 88 (MyD88) and TIR-domain-containing adapter-inducing interferon-β (TRIF), the two major TLR adaptor molecules, are activated in a chronic ischemic hypoperfusion model induced by occlusion of the common carotid artery and also in ischemic models in other organs24,25,26. However, these are not activated in the focal ischemia induced by MCAO or in clinical stroke2,9,27. Why might such a difference exist? In this context, we should consider the following possibilities regarding TLR4 signaling pathway in focal ischemia. First, consider the genetic inhibition of transforming growth factor β–activated kinase 1 (TAK1). Although short-term inhibition by its selective inhibitor leads to protection, such inhibition does not have overt protective effects against cerebral ischemia because of the emergence of an alternative pathway20. This suggests that inhibition of TLR adapters by genetic may lead to up-regulation of another pathway and consequent compensation for the original inhibition. Secondly, TLR2 has been reported to play a critical role in focal cerebral ischemia, like TLR428,29,30. However, TLR2 deficiency reportedly does not protect against cerebral ischemia and treatment with its ligand actually leads to a significant reduction, not exacerbation, of ischemic injury31,32. These findings indicate that the different roles played by TLR2 and TLR4 may responsible for the above unclear role of MyD88 in cerebral ischemia, since this pathway is activated by both receptors2,33. Furthermore, it is possible that different down-stream adaptors are involved during TLR4 signaling in cerebral ischemia. Here, we have shown an association of TLR4 with NOX4 in our model of cerebral ischemia. Indeed, we found that NOX4 was up-regulated and co-localized with TLR4, which was also up-regulated, not only after ischemia and reperfusion in mice, but also in human stroke patient. In addition, NOX4 was co-immunoprecipitated with TLR4 after ischemia in mice. NOX4 has been shown to be a constitutive ROS-generating enzyme that requires the membrane-associated subunit p22phox, but unlike NOX1, NOX2, or NOX3 it does not require the presence of organizers NOXAs or NOXOs subunits16. NOX4 is thought to be an inducible NOX isoform regulated at the mRNA level34. In the present study, inhibiting TLR4 signaling, either genetically or pharmacologically, led to suppress NOX4 induction and reduced oxidative/nitrative stress. Taken together, these findings indicate that NOX4 is activated through TLR4 and mediates ROS production, resulting in a deterioration of the ischemic injury (Fig. 6). This notion is consistent with a report that inhibition of NOX4, but not of either NOX1 or NOX2, largely protects against cerebral ischemia15.
The number of necrotic cells present after cerebral ischemia is considerable and danger-signal molecules are released from such necrotic cells1. One of these molecules is HMGB1, which diffuses out of the necrotic neuronal cells in the ischemic brain and activates various types of cells, such as neurons, glia and endothelial cells1. However, HMGB1 release occurs in the hyperacute phase and it disappears rapidly1, so the explanation for its receptors, such as TLR4 and TLR2, still being activated in the acute phase of cerebral ischemia remains unknown4,7. A recent report has demonstrated that extracellular peroxiredoxin, which is anti-oxidant enzyme itself, released from the ischemic core region acts as a danger signal through TLR2 and TLR4, especially in the acute phase of cerebral ischemia35. These findings indicate the importance of inhibiting TLR4, which reacts with various ligands because of its characteristic of pattern recognition, not only in the hyperacute phase but also in the acute phase, after stroke. Other presently unknown TLR4 ligands may also affect the outcome and inhibiting TLR4 stimulated by all TLR4 ligands may be a strong strategy against stroke. Hence, inhibition of TLR4, leading to termination of the persistent inflammation resulting from recognizing self-components as non-self, has great potential as a strategy against cerebral ischemia.
In conclusion, we have shown that pharmacological inhibition of TLR4 after cerebral ischemia can prevent the progression of the ischemic insults. Furthermore, we have also shown that TLR4-NOX4 signal-mediated ROS production may contribute to the neuronal damage induced by ischemia and reperfusion. These findings indicate that inhibiting TLR4-NOX4 signaling is a promising candidate for a treatment of cerebral ischemia.
Methods
Animals
The experimental designs and all procedures were in accordance with the guidelines of the World Medical Association’s Declaration of Helsinki and the U.S. Department of Health and Human Services Guide for the Care and Use of Laboratory Animals and permission for the study was granted by the Experimental Committee of Gifu Pharmaceutical University. Mice were randomized and the scientists were blinded to group. All efforts were made to minimize both suffering and the number of animals used. Pharmacological experiments were performed using male ddY mice aged 4–5weeks (Japan SLC Ltd., Shizuoka, Japan). TLR4 knock-out (KO) mice were obtained from Dr. Shizuo Akira and Dr. Satoshi Uematsu (Department of Host Defense, Research Institute for Microbial Diseases, Osaka University, Osaka, Japan)36 and backcrossed with C57BL/6 for nine interbreeding generations. Age-matched 8–12 weeks WT C57BL/6 mice were used. The animals (weighing 22 to 28 g) were housed at 24 ± 2°C under a 12-h light/dark cycle (lights on from 07:00–19:00 h). Each animal was used for one experiment only.
Focal cerebral ischemia model in mice
The filament middle cerebral artery occlusion (MCAO) model was used, as described previously7. Anesthesia was induced using 2.0 to 3.0% isoflurane (Merck Hoei Ltd., Osaka, Japan) and maintained using 1.0 to 1.5% isoflurane (both in 70% N2O/30% O2) by means of an animal general anesthesia machine (Soft Lander; Sin-ei Industry Co. Ltd., Saitama, Japan). Body temperature was maintained at 37.0–37.5°C with the aid of a heating pad and heating lamp. After a midline skin incision, the left external carotid artery was exposed and its branches were occluded37. An 8–0 nylon monofilament (Ethicon, Somerville, NJ, USA) coated with a mixture of silicone resin (Xantopren; Bayer Dental, Osaka, Japan) was introduced into the left internal carotid artery through the external carotid artery stump so as to occlude the origin of the middle cerebral artery. Then, the left common carotid artery was occluded. After 2 h of occlusion, the animal was reanesthetized briefly and reperfusion initiated via withdrawal of the monofilament. Just after reperfusion, resatorvid or vehicle was injected intracerebroventricularly. After the surgery, the mice were kept in the preoperative condition (room temperature; 24 ± 2°C) until sampling.
Drug treatment
Resatorvid, {ethyl (6R)-6-[N-(2-chloro-4-fluorophenyl)sulfamoyl]cyclohex-1-ene-1- carboxylate (Fig. 1A)} was synthesized using a procedure based on a previous report38, at laboratory of Pharmaceutical and Medicinal Chemistry, Department of Organic and Medicinal Chemistry, Gifu Pharmaceutical University. The compound was identified by 1H-NMR and Mass spectra (see Supplementary Information.) It was dissolved in 10% dimethyl sulfoxide (DMSO; Nacalai Tesque, Kyoto, Japan), with 10% DMSO in saline being used as vehicle. Resatorvid 1 μl (at 0.001, 0.03, or 0.01 μg/μl) was administered intracerebroventricularly (i.c.v.) at 2 h after ischemia. Just after reperfusion, a Hamilton syringe was used to give each mouse intracerebroventricularly (i.c.v.)39 with 0.001, 0.03 and 0.01 μg of resatorvid in 2 μl of 10% DMSO. Vehicle-treated control mice were injected with the same volume of 10% DMSO.
Physiological monitoring
A polyethylene catheter inserted into the left femoral artery was used to measure arterial blood pressure and heart rate (Power Lab/ 8SP; AD Instrument, Osaka, Japan) at 20 min before and 30 min after MCAO. Blood samples (50 μl) were taken before and at 30 min after the onset of ischemia for pharmacokinetic analysis, pH, pCO2 and pO2 being measured (i-STAT 300F; Abbot Co., Abbot Park, IL, USA). Regional cerebral blood flow (rCBF) was monitored by Doppler flowmetry (Omegaflow flo-N1; Omegawave Inc., Tokyo, Japan). A flexible probe was fixed to the skull (2 mm posterior and 6 mm lateral to bregma). Physiologic monitoring was carried out separately from the main study.
Assessment of cerebral infarction
To analyze infarct volume, mice were euthanized using sodium pentobarbital (Nissan Kagaku, Tokyo, Japan) at 24 h after MCAO and forebrains were coronally sectioned into five slices (2 mm thick). These were placed in 2% 2,3,5-triphenyltetrazolium chloride (TTC; Sigma-Aldrich Co., St. Louis, MO, USA) at 37°C for 30 min and then fixed in 10% buffered formalin. Digital images of the caudal aspect of each slice were obtained using a digital camera (Coolpix 4500, Nikon, Tokyo, Japan). Infarct, ipsilateral hemisphere and contralateral hemispere areas were measured using image processing software (Image-J ver. 1.43 h; National Institutes of Health, Bethesda, MD, USA) and infarct volume was calculated as previously reported40.
Neurological deficits
Mice were tested for neurological deficits at 24 h after ischemia and reperfusion. Scoring was done as described previously37, using the following scale: 0, no observable neurological deficits (normal); 1, failure to extend the right forepaw (mild); 2, circling to the contralateral side (moderate); 3, loss of walking or righting reflex (severe). The investigator who rated the mice was masked as to the group to which each mouse belonged.
Nuclear and cytoplasmic extraction
Whole brains were cut to provide one 3-mm coronal section each (between 5 and 8 mm from the frontal extent of the forebrain), then carefully separated into ipsilateral and contralateral sides. From the separated sections, nuclear and cytoplasmic fractions were obtained with the aid of a nuclear extraction kit (Trans AM; Active Motif, Carlsbad, CA, USA). Assays to determine protein concentrations were performed using a BCA protein assay kit (Pierce Biotechnology, Rockford, IL, USA). An aliquot of 5 µg of protein from the nuclear or cytoplasmic fraction was subjected to 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then the separated proteins were transferred onto a polyvinylidenedifluoride membrane. For immunoblotting, monoclonal anti-p65 antibody (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and monoclonal anti-IRF-3 antibody (1:1000; Cell Signaling Technology, Danvers, MA, USA) were used. The secondary antibodies were anti-mouse HRP-conjugated IgG (1:2000; Pierce Biotechnology, IL, USA) and anti-rabbit HRP-conjugated IgG (1:2000; Pierce Biotechnology). The immunoreactive bands were visualized using ImmunoStar LD (Wako Pure Chemical Industries, Osaka, Japan). The band intensity was measured using a Lumino imaging analyzer (LAS-4000: Fuji Film, Tokyo, Japan). The differences of expression level were analyzed using MultiGauge software (Fujifilm) by measuring the intensity of the bands. Histone H1 and β-actin were used as the internal controls for the nuclear and cytoplasmic fractions, respectively.
Western blotting and immunoprecipitation
Mice were deeply anesthetized and decapitated at 24 h after ischemia. Brains were quickly removed and the brains were cut into 3 mm coronal sections and separated into ipsilateral side and contralateral side. Tissues were homogenized in 10 ml/g tissue ice-cold lysis buffer (50 mM Tris-HCl, pH 8.0, containing 150 mM NaCl, 50 mM EDTA, 1% Triton X-100 and protease/phosphatase inhibitor mixture). Immunoprecipitation was performed using Classic IP Kit (Thermo Fisher Scientific, Inc., Waltham, MA, USA). A sample obtained from the sham-control or ischemic hemisphere was loaded and the separated proteins were subsequently transferred. For immunoblotting, the following primary antibodies were used: polyclonal anti-phospho-p38 antibody (1:2000; Promega, Madison, WI, USA), polyclonal anti-p38 antibody (1:1000; Cell Signalimg Technology), monoclonal anti-phospho-c-jun antibody (1:1000; Cell Signaling Technology), monoclonal anti-c-jun antibody (1:1000; Cell Signaling Technology), polyclonal anti-MMP9 antibody (1:1000; Millipore, Billerica, MA, USA) and polyclonal anti-NOX4 antibody (1:500; Abcam, Cambridge, UK). Using fresh samples of the sham-operated or ischemic hemisphere, the association of TLR4 with NOX4 was examined by immunoprecipitation with monoclonal anti-TLR4 antibody (1:20; Imgenex, San Diego, CA, USA) followed by immunoblotting with polyclonal anti-NOX4 antibody (1:500; Abcam). The secondary antibody was anti-rabbit HRP-conjugated IgG (1:2000; Pierce Biotechnology). The immunoreactive bands were visualized using ImmunoStar LD (Wako Pure Chemical Industries). The band intensity was measured using a Lumino imaging analyzer (LAS-4000: Fuji Film). The differences of expression level were analyzed using MultiGauge software (Fujifilm) by measuring the intensity of the bands.
Immunohistochemistry for oxidation and nitration
Sham-operated or ischemic mice were injected with sodium pentobarbital (Nembutal; 50 mg/kg, i.p.), then perfused through the left ventricle with 4% paraformaldehyde in 0.1 M phosphate buffer (PB; pH 7.4). Brains were removed after 15 min perfusion fixation at 4°C, then immersed in the same fixative solution overnight at 4°C. They were then immersed in 25% sucrose in 0.1 M PB for 24 h and finally frozen in liquid nitrogen. Coronal sections (14 μm thick) between from 0.4 and 1.0 mm anterior to bregma were cut on a cryostat at −20°C and stored at −80°C until use. The sections were blocked with 10% goat serum in PBS, then incubated overnight at 4°C with the following primary antibodies: polyclonal anti-8-OHdG antibody (1:20; JalCA, Fukuroi, Shizuoka, Japan) or monoclonal anti-nitrotyrosine antibody (1:100; Cayman Chemical, Ann Arbor, MI, USA). Then, they were incubated for 3 h with Alexa Fluor 546 F (ab’)2 fragment of goat anti-mouse IgG (H+L) antibody. To standardize the measurements, two predesignated squares in the peri-infarct region of the cortex were counted and averaged. The area of peri-infarct reagion was defined as previously reported41.
Double-immunostaining
For double-immunostaining, frozen tissues from mice and paraffin-embedded human specimens were used. The human specimens were deparaffinized and rehydrated before the immunohistochemical procedures. Coronal sections from mice and human brain slices were incubated overnight at 4°C with monoclonal anti-TLR4 antibody (1:100; Imgenex) and polyclonal anti-NOX4 antibody (1:100; Abcam). The secondary antibodies were Alexa Fluor 546 F (ab’)2 fragment of goat anti-mouse IgG (H+L) antibody and Alexa Fluor 488 F (ab’)2 fragment of goat anti-rabbit IgG (H+L) antibody. The sections were observed under a confocal microscope (FV10i, Olympus, Tokyo, Japan).
Human tissue specimens
Human tissue specimens were obtained from patients who had undergone surgery for reasons of clinical necessity at the Department of Neurosurgery, Gifu University Hospital. There were no additional interventions in the patients enrolled in this study. The use of surgical specimens for immunohistochemistry was approved by the institutional review board of Gifu University (#24–130) and all patients or their representative signed informed written consent. The stroke patient was a woman of 60’s who suffered large hemispheric infarction due to cardiogenic embolism and underwent internal and external decompression at 24 h after symptom onset because of brain herniation. The tissue specimen was temporal cortex of the infarct brain removed by decompressive surgery. The control patient was a woman of 20’s who presented intractable epilepsy caused by cavernous malformation in the temporal lobe and had no other medical history and the neurological status was completely normal with the exception of seizures. The patient underwent the anterior temporal lobectomy including the removal of vascular malformation. The tissue specimen was the histologically normal cortex of the removed temporal lobe.
Neuronal apoptosis
Frozen samples from mice were used. Coronal sections (14 μm thick) between from 0.4 and 1.0 mm anterior to bregma were cut on a cryostat at −20°C and stored at −80°C until use. To qualify the number of apoptotic neuronal cells after MCAO, we counted the number of cells in which positivity for Hoechst33342 (1:1000; Invitrogen, Carlsbad, CA, USA), a nuclear marker, was co-located with cleaved caspase-3 (1:400; Cell Signaling Technology), an apoptosis marker and Neuronal Nuclei (NeuN) (1:1000; Chemicon, Temecula, CA, USA), a neuronal marker. The secondary antibodies were Alexa Fluor 546 F (ab’)2 fragment of goat anti-mouse IgG (H+L) antibody and Alexa Fluor 488 F (ab’)2 fragment of goat anti-rabbit IgG (H+L) antibody. The sections were observed under a confocal microscope (FV10i, Olympus, Tokyo, Japan) and the number of apoptotic neuronal cells were counted using image processing software (Image-J ver. 1.43 h; National Institutes of Health). To standardize the measurements, two predesignated squares in the peri-infarct region of the cortex were counted and averaged.
Statistical analysis
All data are presented as means ± standard deviation. Student two-tailed t-test was used for comparisons of two experimental groups and one-way ANOVA followed by Dunnett test was used for multiple group comparisons. The Mann-Whitney U-test was used for the statistical analysis of neurological deficits. Stat View software version 5.0 (SAS Institute Inc., Cary, NC, USA) was used and P<0.05 was considered statistically significant.
References
Qiu, J. et al. Early release of HMGB-1 from neurons after the onset of brain ischemia. J Cereb Blood Flow Metab 28, 927–938 (2008).
Yang, Q. W. et al. Upregulated expression of toll-like receptor 4 in monocytes correlates with severity of acute cerebral infarction. J Cereb Blood Flow Metab 28, 1588–1596 (2008).
Cao, C. X. et al. Reduced cerebral ischemia-reperfusion injury in Toll-like receptor 4 deficient mice. Biochem Biophys Res Commun 353, 509–514 (2007).
Tang, S. C. et al. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci U S A 104, 13798–13803 (2007).
Caso, J. R. et al. Toll-like receptor 4 is involved in brain damage and inflammation after experimental stroke. Circulation 115, 1599–1608 (2007).
Caso, J. R. et al. Toll-like receptor 4 is involved in subacute stress-induced neuroinflammation and in the worsening of experimental stroke. Stroke 39, 1314–1320 (2008).
Hyakkoku, K. et al. Toll-like receptor 4 (TLR4), but not TLR3 or TLR9, knock-out mice have neuroprotective effects against focal cerebral ischemia. Neuroscience 171, 258–267 (2010).
Qiu, J. et al. High-mobility group box 1 promotes metalloproteinase-9 upregulation through Toll-like receptor 4 after cerebral ischemia. Stroke 41, 2077–2082 (2010).
Yang, Q. W. et al. HMBG1 mediates ischemia-reperfusion injury by TRIF-adaptor independent Toll-like receptor 4 signaling. J Cereb Blood Flow Metab 31, 593–605 (2011).
Chan, P. H. Role of oxidants in ischemic brain damage. Stroke 27, 1124–1129 (1996).
Chan, P. H. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab 21, 2–14 (2001).
Eliasson, M. J. et al. Neuronal nitric oxide synthase activation and peroxynitrite formation in ischemic stroke linked to neural damage. J Neurosci 19, 5910–5918 (1999).
Steinhubl. Why have antioxidants failed in clinical trials? Am J Cardiol 101, 14–19 (2008).
Chen, H., Song, Y. S. & Chan, P. H. Inhibition of NADPH oxidase is neuroprotective after ischemia-reperfusion. J Cereb Blood Flow Metab 29, 1262–1272 (2009).
Kleinschnitz, C. et al. Post-stroke inhibition of induced NADPH oxidase type 4 prevents oxidative stress and neurodegeneration. PLoS Biol 8, pii e1000479 (2010).
Lambeth, J. D. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol 4, 181–189 (2004).
Park, H. S. et al. Cutting edge: direct interaction of TLR4 with NAD(P)H oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-kappa B. J Immunol 173, 3589–3593 (2004).
Ben, M. S. et al. Heat shock protein gp96 and NAD(P)H oxidase 4 play key roles in Toll-like receptor 4-activated apoptosis during renal ischemia/reperfusion injury. Cell Death Differ 17, 1474–1485 (2010).
Sha, T. et al. Therapeutic effects of TAK-242, a novel selective Toll-like receptor 4 signal transduction inhibitor, in mouse endotoxin shock model. Eur J Pharmacol 57, 231–239 (2007).
Neubert, M., Ridder, D. A., Bargiotas, P., Akira, S. & Schwaninger, M. Acute inhibition of TAK1 protects against neuronal death in cerebral ischemia. Cell Death Differ 18, 1521–1530 (2011).
Asahi, M. et al. Role for matrix metalloproteinase 9 after focal cerebral ischemia: effects of gene knockout and enzyme inhibition with BB-94. J Cereb Blood Flow Metab 20, 1681–1689 (2000).
Grossetete, M. & Rosenberg, G. A. Matrix metalloproteinase inhibition facilitates cell death in intracerebral hemorrhage in mouse. J Cereb Blood Flow Metab 28, 752–763 (2008).
Takashima, K. et al. Analysis of binding site for the novel small-molecule TLR4 signal transduction inhibitor TAK-242 and its therapeutic effect on mouse sepsis model. Br J Pharmacol 157, 1250–1262 (2009).
Gao, Y., Fang, X., Tong, Y., Liu, Y. & Zhang, B. TLR4-mediated MyD88-dependent signaling pathway is activated by cerebral ischemia-reperfusion in cortex in mice. Biomed Pharmacother 63, 442–450 (2009).
Hua, F. et al. Blocking the MyD88-dependent pathway protects the myocardium from ischemia/reperfusion injury in rat hearts. Biochem Biophys Res Commun 338, 1118–1125 (2005).
Tsung, A. et al. The transcription factor interferon regulatory factor-1 mediates liver damage during ischemia-reperfusion injury. Am J Physiol Gastrointest Liver Physiol 290, 1261–1268 (2006).
Hua, F. et al. The TRIF-dependent signaling pathway is not required for acute cerebral ischemia/reperfusion injury in mice. Biochem Biophys Res Commun 390, 678–683 (2009).
Ziegler, G. et al. TLR2 has a detrimental role in mouse transient focal cerebral ischemia. BiochemBiophys Res Commun 359, 574–579 (2007).
Ziegler, G. et al. Blocking TLR2 in vivo protects against accumulation of inflammatory cells and neuronal injury in experimental stroke. J Cereb Blood Flow Metab 31, 757–766 (2011).
Brea, D. et al. Toll-like receptors 2 and 4 in ischemic stroke: outcome and therapeutic values. J Cereb Blood Flow Metab 31, 1424–1431 (2011).
Hua, F. et al. Differential roles of TLR2 and TLR4 in acute focal cerebral ischemia/reperfusion injury in mice. Brain Res 1262, 100–108 (2009).
Lu, C. et al. TLR2 ligand induces protection against cerebral ischemia/reperfusion injury via activation of phosphoinositide 3-kinase/Akt signaling. J Immunol 187, 1458–1466 (2011).
Famakin, B. M. et al. Disruption of downstream MyD88 or TRIF Toll-like receptor signaling does not protect against cerebral ischemia. Brain Res 1388, 148–156 (2011).
Martyn, K. D., Frederick, L. M., von, L. K., Dinauer, M. C. & Knaus, U. G. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell Signal 18, 69–82 (2006).
Shichita, T. et al. Peroxiredoxin family proteins are key initiators of post-ischemic inflammation in the brain. Nat Med doi: 10, 1038/nm. 2749 (2012).
Hoshino, K. et al. Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol 162, 3749–3752 (1999).
Hara, H. et al. Attenuation of transient focal cerebral ischemic injury in transgenic mice expressing a mutant ICE inhibitory protein. J Cereb Blood Flow Metab 17, 370–375 (1997).
Yamada, M. et al. Optically Active Cyclohexene Derivative as a New Antisepsis Agent: An Efficient Synthesis of Ethyl (6R)-6-[N-(2-Chloro-4-fluorophenyl) sulfamoyl] cyclohex-1-ene-1-carboxylate (TAK-242). Chem Pharm Bull 54, 58–62 (2006).
Haley, T. J. & Mccormick, W. G. Pharmacological effects produced by intracerebral injection of drugs in the conscious mouse. Br J Pharmacol Chemother 12, 12–15 (1957).
Hara, H. et al. Inhibition of interleukin 1beta converting enzyme family proteases reduces ischemic and excitotoxic neuronal damage. Proc Natl Acad Sci U S A 94, 2007–2012 (1997).
Oida, Y. et al. Post-treatment of a BiP inducer prevents cell death after middle cerebral artery occlusion in mice. Neurosci Lett 484, 43–46 (2010).
Acknowledgements
We thank Dr. Shizuo Akira and Dr. Satoshi Uematsu, Department of Host Defense, Research Institute for Microbial Diseases, Osaka University, Japan, for providing us with TLR4 KO mice. We also thank Mr. Kenjirou Ogawa, Molecular Pharmacology, Department of Biofunctional Evaluation, Gifu Pharmaceutical University, Japan, for technical assistance.
Author information
Authors and Affiliations
Contributions
Y.S., J.H., K.M., K.T., M.S. and H.H. designed the experiments. Y.S. and J.H. performed the experiments. K.H., T.M. and H.N. contributed the synthesis of resatorvid. Y.E., M.I., Y.H., S.Y. and T.I. contributed the clinical samples. H.T. and N.I. contributed the TLR4 KO mice. Y.S. performed the analysis and wrote the paper. All authors contributed to the editing of the paper and to scientific discussions.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Suzuki, Y., Hattori, K., Hamanaka, J. et al. Pharmacological inhibition of TLR4-NOX4 signal protects against neuronal death in transient focal ischemia. Sci Rep 2, 896 (2012). https://doi.org/10.1038/srep00896
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep00896
This article is cited by
-
TRIM-containing 44 aggravates cardiac hypertrophy via TLR4/NOX4-induced ferroptosis
Journal of Molecular Medicine (2023)
-
Crosstalk Between Autophagy and Inflammation in Chronic Cerebral Ischaemia
Cellular and Molecular Neurobiology (2023)
-
Brain vulnerability and viability after ischaemia
Nature Reviews Neuroscience (2021)
-
Attenuation of the Induction of TLRs 2 and 4 Mitigates Inflammation and Promotes Neurological Recovery After Focal Cerebral Ischemia
Translational Stroke Research (2021)
-
Engagement of MicroRNA-155 in Exaggerated Oxidative Stress Signal and TRPA1 in the Dorsal Horn of the Spinal Cord and Neuropathic Pain During Chemotherapeutic Oxaliplatin
Neurotoxicity Research (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
