
Abstract
Cryptococcus neoformans is an opportunistic pulmonary fungal pathogen that disseminates to the CNS causing fatal meningitis in immunocompromised patients. Dendritic cells (DCs) phagocytose C. neoformans following inhalation. Following uptake, cryptococci translocate to the DC lysosomal compartment and are killed by oxidative and non-oxidative mechanisms. DC lysosomal extracts kill cryptococci in vitro; however, the means of antifungal activity remain unknown. Our studies determined non-oxidative antifungal activity by DC lysosomal extract. We examined DC lysosomal killing of cryptococcal strains, anti-fungal activity of purified lysosomal enzymes and mechanisms of killing against C. neoformans. Results confirmed DC lysosome fungicidal activity against all cryptococcal serotypes. Purified lysosomal enzymes, specifically cathepsin B, inhibited cryptococcal growth. Interestingly, cathepsin B combined with its enzymatic inhibitors led to enhanced cryptococcal killing. Electron microscopy revealed structural changes and ruptured cryptococcal cell walls following treatment. Finally, additional studies demonstrated that osmotic lysis was responsible for cryptococcal death.
Similar content being viewed by others


Introduction
Cryptococcus neoformans is a ubiquitous encapsulated opportunistic fungal pathogen that causes pneumonia and meningitis in immunocompromised patients, especially in recipients of organ transplants or individuals with AIDS1,2,3,4. The infection is acquired through the inhalation of desiccated yeast mostly from soil contaminated with pigeon droppings1. Although C. neoformans is most often acquired via the pulmonary route, few patients present with a clinical picture of pneumonia (reviewed in1,2,5). The predominant protective immune response to C. neoformans is cell-mediated immunity (CMI) primarily by Th1-type CD4+ T cells6,7,8,9,10,11. However, the first line of defense against C. neoformans is the innate immune system. The adaptive immune system must be primed by innate antigen-presenting cells (APCs), most importantly dendritic cells (DCs), as DCs are the predominant APC capable of antigen presentation to naïve T cells12. DCs are considered sentinels of the immune system13 and can be especially important in surveillance at mucosal sites, such as the lung14. DCs phagocytose, degrade and process extracellular foreign organisms for antigen presentation primarily on class II MHC to CD4+ T helper cells to initiate the adaptive immune response15,16. In addition to their role as APCs, DCs and other innate cells can be involved in direct microbicidal activity against pathogens17,18,19. DCs have been shown to be involved in killing C. neoformans as well as other pathogenic fungi19,20,21,22,23.
In vitro, DCs phagocytose C. neoformans following opsonization with complement or antibody20,23 and the organism can be killed by both oxidative and non-oxidative mechanisms23. In vivo studies showed that C. neoformans can be phagocytosed by murine pulmonary DCs24. After phagocytosis by DCs, C. neoformans enters the endosomal compartment and traffics to the lysosomal compartment, where it is degraded by the lysosomal components and processed for antigen presentation20. Following cryptococcal uptake and processing, DC maturation is evident by increased surface expression of MHC II and the costimulatory molecules CD80 and CD86. These DCs are then able to present antigen to cryptococcal-specific T cells and induce T cell proliferation24. Crude lysosomal extracts generated from murine bone-marrow-derived DCs (BMDCs) exhibit anticryptococcal activity in vitro in a dose-dependent manner20.
Lysosomes are cellular organelles involved in the degradation of phagocytosed/endocytosed and intracellular material25. They are the site of MHC class II loading and are important sites for killing and degradation of phagocytosed material, including pathogens25,26. They contain hydrolases that degrade endocytosed material27. DC lysosomes contain an assortment of endopeptidases, exopeptidases, estearases and reductases26,28,29,30, but the majority of the proteases are the cysteine proteases such as cathepsin B, L and S and the aspartate proteases cathepsin D and E26,28,29,30. Other cysteine proteases are also present in lysosomes but are more closely related to the caspases25. Cathepsins D and L act as endoproteases and cathepsin B has poor endoproteolytic activity but is a strong carboxypeptidase; nonetheless, each has a role in antigen processing28,31,32. Cathepsin L-deficient mice cannot process the invariant chain (Ii) and have defects in MHC II antigen presentation25. Cathepsins B and D also play a role in Ii processing and MHC II presentation25,27. Because of their importance in antigen degradation and processing, we primarily focused on the cathepsins as potential antifungal agents. In addition to enzymes, lysosomes also contain small peptides called antimicrobial peptides (AMPs), such as α- and β-defensins and the cathelicidins33. AMPs work by causing rupture of the membranes of bacteria, viruses, parasites and fungi, or by interfering with DNA and RNA33,34,35.
In the present study, we determined DC lysosomal components that may play a role in the killing of Cryptococcus. Accordingly, we examined the antifungal activity of the DC lysosomal extract against four serotypes of Cryptococcus, tested the ability of purified lysosomal cathepsins to kill the organism and blocked enzymatic activity of the cathepsins that demonstrated anti-cryptococcal activity. Finally, we investigated mechanisms of anti-cryptococcal activity by these lysosomal components.
Results
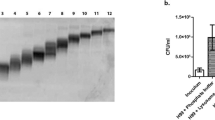
DC-derived lysosomal extract kills Cryptococcus strains in vitro
Previous studies have shown that DC-derived lysosomal extract can kill C. neoformans serotype A strain 145 in vitro20. To confirm DC lysosomal activity against other cryptococcal strains and serotypes, C. neoformans serotype A strain H99, C. gattii serotype B strain R265, C. gattii serotype C strain WSA87 and C. neoformans serotype D strain R4249 yeast cells were individually incubated in phosphate buffer alone or in phosphate buffer with lysosomal extract for 24 h at 37°C, following which the CFU in the wells were determined. We found that the DC-derived lysosomal extracts have significant antifungal activity against all cryptococcal strains tested, leading to complete killing of each strain of Cryptococcus (Figure 1). Because all strains and serotypes were equally affected, further studies were conducted using the highly pathogenic C. neoformans strain H99.

DC-derived lysosomal extract kills Cryptococcus in vitro.
C. neoformans serotype A strain H99, C. gattii serotype B strain R265, C. gattii serotype C strain WSA87 and C. neoformans serotype D strain R4249 yeast cells at 2.5 x 105 cells/ml were incubated in phosphate buffer alone or in phosphate buffer with lysosomal extract for 24 h at 37°C and CFU in the wells determined as described in Materials and Methods. White bars indicate inoculum, gray bars indicate cryptococcal cells incubated in phosphate buffer alone and black bars indicate cryptococcal cells incubated with lysosomal extract. Data shown are means ± standard errors of the means (SEM) of the cumulative results of three independent experiments. An asterisk * indicates a significant difference compared to the results for the yeast incubated in phosphate buffer alone (P < 0.0001) and τ indicates a significant difference compared to the results for inocula (P < 0.0001).
Purified lysosomal enzymes inhibit cryptococcal growth in vitro
Many lysosomal components of DCs can contribute to killing and degradation of phagocytosed organisms. Lysosomal enzymes, such as cathepsins, are known to be involved in intracellular degradation and processing for antigen presentation36,37. Therefore, we chose to examine the role of cathepsins in the killing of C. neoformans strain H99. In order to examine the role of cathepsins in cryptococcal killing or inhibition, C. neoformans strain H99 yeast cells were incubated in phosphate buffer with 10 µg/ml or 50 µg/ml of cathepsin B (Figure 2A), cathepsin D (Figure 2B), or cathepsin L (Figure 2C) for 24 h at 37°C, following which the CFUs were determined. These concentrations were chosen to represent physiological concentration found in cell lysates (10 µg)38,39 and excess concentration (50 µg). Results showed that cathepsin B significantly inhibited the growth of C. neoformans at 10 µg/ml and at 50 µg/ml showed even greater inhibition almost to inocula levels (Figure 2A). Incubation of C. neoformans with cathepsin D at 10 µg/ml and 50 µg/ml had no effect on the growth when compared to growth in the phosphate buffer alone (Figure 2B). Cathepsin L inhibited the growth of C. neoformans at both 10 µg/ml and 50 µg/ml compared to the growth in the phosphate buffer alone, but this was not a statistically significant difference (Figure 2C). Because the only significant inhibition of C. neoformans growth was observed following incubation with cathepsin B, further studies focused only on cathepsin B.

Purified lysosomal enzymes inhibit C. neoformans growth in vitro.
C. neoformans strain H99 yeast cells were incubated in phosphate buffer with 10 µg/ml or 50 µg/ml of cathepsin B (A), cathepsin D (B), or cathepsin L (C) for 24 h at 37°C, following which the CFU in the wells were determined as described in Materials and Methods. White bars indicate inoculum, gray bars indicate cryptococcal cells incubated in phosphate buffer, hashed bars indicate cryptococcal cells incubated with 10 µg/ml cathepsin and black bars indicate cryptococcal cells incubated with 50 µg/ml cathepsin. Data shown are means ± standard errors of the means (SEM) of the cumulative results of 3 independent experiments. An asterisk indicates a significant difference compared to the results for H99 incubated in phosphate buffer alone (p < 0.05).
Confirmation of the presence of cathepsin B in BMDC lysosomal extracts
In order to definitively confirm that cathepsin B was present in our DC lysosomal extract, the extract was examined for the presence of cathepsin B by Western blot analysis. Proteins from two separate preparations of DC lysosomal extract were separated by SDS-PAGE and transferred to a PVDF membrane. Blots were probed with anti-human/mouse cathepsin B monoclonal antibody. Recombinant mouse cathepsin B in the pro-form (39 kDa), was used as a positive control. Active cathepsin B is approximately 25–26 kDa40. The Western blot showed the presence of cathepsin B in two separate preparations of BMDC lysosomal extract (Figure 3A). We next determined the concentration of cathepsin B in the BMDC lysosomal extracts by ELISA. Aliquots of each step during the lysosomal purification process were examined to quantify the amount of cathepsin B present. The ELISA showed that an average amount of 4000 pg/mg protein of cathepsin B is present in BMDC lysosomal extracts (Figure 3B).

Confirmation of the presence of Cathepsin B in BMDC lysosomal extracts.
Lysosomal extracts prepared from BMDCs were examined for the presence of cathepsin B. For ELISA analysis, fractions were taken during each step of the lysosomal purification process to examine the amount of cathepsin B present. Recombinant mouse cathepsin B in the pro-form was used as the positive control for Western blot analysis and for the ELISA standard. Cathepsin B was detected by separating the lysosomal extracts through SDS-page then transferring to a PVDF membrane and probing with anti-human/mouse cathepsin B. Recombinant mouse cathepsin B in the pro-form is 39 kDa and active cathepsin B is 25-26 kDa. A). Western blot analysis of cathepsin B shows the presence of cathepsin B in two different samples of BMDC lysosomal extract. The blot is representative of three separate experiments. B) ELISA shows that 4000 pg cathepsin B per mg protein is present in BMDC lysosomal extracts. Aliquots of the different stages in the lysosomal purification process showed that cathepsin B is found primarily in the lysosomal fraction. Data shown are means ± standard error of the mean (SEM) of three independent experiments.
Enzymatic inhibition of cathepsin B activity does not affect its ability to inhibit the growth of C. neoformans
Given our observations that the 50 µg/ml concentration of cathepsin B significantly inhibited the growth of C. neoformans strain H99 when compared to the growth in the phosphate buffer alone, we determined whether the inhibition of growth could be reversed by inhibiting the enzymatic activity of cathepsin B. For this, C. neoformans strain H99 yeast cells were incubated in phosphate buffer alone, in phosphate buffer with either cathepsin B, an inhibitor, or cathepsin B plus an inhibitor for 24 h at 37°C, following which the CFUs were determined. The inhibitors used were calpain inhibitor at 10 µg/ml, cathepsin B & L inhibitor at 50 or 100 µM and CA-074 at 20 µM. Calpain inhibitor is only soluble in DMSO, which is toxic to C. neoformans at higher concentrations, so higher concentrations of the calpain inhibitor could not be used in our assay (data not shown). CA-074 is a specific inhibitor of only cathepsin B, cathepsin B&L inhibitor inhibits cathepsin B and L only and calpain is a general inhibitor of cathepsin activity. However, because our assays use only purified cathepsin B and not the entire DC lysosomal extract, the specificity of the inhibitors is not a factor. Surprisingly, our results showed that the addition of any cathepsin B inhibitor, at the concentrations used, enhanced the antifungal activity of cathepsin B rather than abrogating its activity (Figure 4A). To determine whether this observation was a result of the failure of the inhibitors to prohibit enzymatic activity of cathepsin B, we measured the percent inhibition of the cathepsin B enzymatic activity with each inhibitor (Figure 4B). We found that each inhibitor did significantly inhibit the enzymatic activity of cathepsin B. The calpain inhibitor showed 47.34% inhibition (52.66% activity) at 10 µg/ml, the cathepsin B&L inhibitor showed 55.67% inhibition (44.33% activity) at 50 µM and 95.83% inhibition (4.13% activity) at 100 µM and CA-074 showed 95.69% inhibition (4.31% activity) at 20 µM, suggesting that cathepsin B may have non-enzymatic activity against C. neoformans.

Inhibition of cathepsin B enzymatic activity enhances the antifungal effects against C. neoformans.
C. neoformans strain H99 yeast cells were incubated in phosphate buffer alone, in phosphate buffer with either 50 µg/ml cathepsin B, 10 µg/ml calpain inhibitor, 50 or 100 µM cathepsin B & L inhibitor, 20 µM CA-074, or cathepsin B plus an inhibitor for 24 h at 37°C, following which the numbers of CFU in the wells were determined. A) Data shown are means ± standard errors of the means (SEM) of the cumulative results of 3 independent experiments. An asterisk * indicates a significant difference compared to the results for H99 in phosphate buffer alone (p < 0.05) and τ indicates a significant difference compared to the initial the inocula (white bar) (p < 0.05). B) The percent cathepsin B enzymatic activity following incubation with each different inhibitor was measured using the Cathepsin B Activity Assay Kit. Data shown are means ± standard errors of the means (SEM) of the cumulative results of 3 independent experiments. Asterisks * show significant differences in activity compared to cathepsin B alone (p < 0.05).
Microscopy of C. neoformans treated with cathepsin B and cathepsin B + inhibitors show cryptococcal structural changes and cell wall rupture
Due to the killing observed following incubation with cathepsin B and its inhibitors, we were interested in determining their effects on the fungus during in vitro incubation. To assess this, scanning electron microscopy (SEM) (Figure 5A) or transmission electron microscopy (TEM) (Figure 5B) was performed on C. neoformans after growth in the presence of phosphate buffer, cathepsin B, calpain inhibitor, CA-074, or cathepsin B + an inhibitor for 12 h. We chose to examine the cells after 12 hours of incubation in order to observe the effects of cathepsin B and either calpain inhibitor or CA-074 on live cells, since 24 h of incubation results in complete killing of C. neoformans. SEM of C. neoformans strain H99 showed structural changes in the organism following incubation with cathepsin B and cathepsin B + calpain inhibitor or CA-074 compared to untreated conditions (Figure 5A). TEM of C. neoformans strain H99 showed a crescent morphology indicative of structural changes and possible cell death41 due to cathepsin B and cathepsin B + calpain inhibitor or CA-074 treatment compared to untreated cells or cells treated with calpain inhibitor or CA-074 alone (Figure 5B). In addition, cells showing structural changes also show ruptured cell walls but intact cell membranes (Figure 5B). Quantification of EM data showed altered cryptococcal cell morphology (crescent-shaped) in 10% of untreated cells and in 17% and 15% of calpain or CA-074 treated cells, respectively. Further, cells treated with cathepsin B, calpain inhibitor + cathepsin B, or CA-074 + cathepsin B had statistically higher percentages of altered cell morphology (27%, 27% and 25%, respectively) compared to cells treated with either untreated cells or compared to cells treated with inhibitors alone (P<0.0001). In support of these data, confocal microscopy using live BMDCs showed that C. neoformans (green) trafficking into DC lysosomal compartments (red) also adopts a similar crescent-shaped morphology (Figure 5C)

Microscopy of C. neoformans treated with cathepsin B and cathepsin B + inhibitors show cryptococcal structural changes.
C. neoformans was grown in the presence of cathepsin B, calpain inhibitor, CA-074, or cathepsin B with inhibitors for 12 h, fixed with 4% formaldehyde/1% glutaraldehyde, prepared for electron microscopy and examined by SEM or TEM. A) SEM of C. neoformans strain H99 shows structural changes (arrows) in the organism following incubation with cathepsin B and cathepsin B + inhibitors, but not in untreated conditions. B) TEM of C. neoformans shows structural changes in cells treated with cathepsin B and cathepsin B + an inhibitor, but not in untreated cells or in those treated with only calpain inhibitor or CA-074. In addition, cells showing structural changes (arrows) also show ruptured cell walls but intact cell membranes. Data shown are representative of 30 fields per condition imaged. Magnification is 5,000 X for SEM images and 25,000 X for TEM images. C) Purified BMDCs were incubated with C. neoformans yeast cells at a 2:1 ratio with 1 µg/ml of Oregon green-labeled 3C2 opsonizing antibody and 50 nM LysoTracker red. Samples were incubated at 37°C for 1 hour prior to imaging. A representative confocal image of C. neoformans organisms (green) shown in a LysoTracker red-positive compartment (red) with a similar crescent-shaped morphology as seen through electron microscopy. Confocal image is representative of 3 experiments performed, 30 fields imaged per experiment.
Cathepsin B and inhibitor treatment result in osmotic lysis of Cryptococcus neoformans
Our results in Figure 5 showed that incubation with cathepsin B and calpain inhibitor lead to a compromised cell wall of C. neoformans. We hypothesized that the cell wall ruptures induced by cathepsin B and inhibitor treatment enhanced osmotic lysis of the cryptococcal cells, resulting in death. To examine this, C. neoformans strain H99 yeast cells were incubated in phosphate buffer alone or in phosphate buffer with 50 µg/ml cathepsin B and 10 µg/ml calpain inhibitor for 24 h at 37°C, following which the CFU in the wells were determined by diluting and plating on YPD agar or osmotically supportive YPD agar containing 1.4 M sorbitol. By plating on the osmotically supportive YPD agar, we almost completely abrogated the killing of C. neoformans (Figure 6). As an alternative approach, electron microscopy (EM) was performed following incubation in YPD + 1.4 M sorbitol broth using cathepsin B with the specific inhibitor CA-074. Both TEM and SEM confirmed that the addition of sorbitol reversed the crescent-shaped appearance of the organism (Figure 6B and 6C). Therefore, we conclude that the cathepsin B plus inhibitor treatment leads to osmotic lysis of the fungus.

Cathepsin B and inhibitor treatment result in osmotic lysis of Cryptococcus neoformans.
A) C. neoformans strain H99 yeast cells were incubated in phosphate buffer alone or in phosphate buffer with 50 µg/ml cathepsin B and 10 µg/ml calpain inhibitor for 24 h at 37°C, following which the CFUs were determined by diluting and plating on YPD agar or osmotically supportive YPD media containing 1.4 M sorbitol. Data shown are means ± standard errors of the means (SEM) of the cumulative results of 3 independent experiments. An asterisk * indicates a significant difference of the cathepsin B and inhibitor treated groups compared to the results for inocula compared (p < 0.05). A τ indicates a significant difference of the cathepsin B and inhibitor treated group grown on sorbitol YPD compared to the results grown on YPD (P < 0.0001). B) C. neoformans strain H99 yeast cells were incubated in phosphate buffer alone or in phosphate buffer with 50 µg/ml cathepsin B and 20 µM CA-074 for 24 h at 37°C, following which the cells were transferred to YPD broth or osmotically supportive YPD broth containing 1.4 M sorbitol for 8 h at 30°C. Following incubation, cells were fixed with 4% formaldehyde/1% glutaraldehyde and processed for electron microscopy. Quantified data (B) are shown from quantification of 30 fields per condition. C) Transmission electron microscopy (TEM) and scanning electron microscopy (SEM) show structural changes in the organism following incubation with CA-074 + cathepsin B (arrows) and abrogation of these changes following incubation in YPD+sorbitol. Data shown are representative of 30 fields per condition imaged. Magnification is 5,000 X for SEM and 25,000 X for TEM images.
Discussion
While many studies have shown that neutrophils and macrophages are able to kill fungal organisms17,42, little is known about the mechanisms of DC killing of fungal organisms. Studies with other pathogenic fungi have shown that there are different outcomes based on which immune cell type encounters the organism. For Histoplasma capsulatum, macrophages are permissive for fungal growth, while DCs kill the organism19. While DCs and macrophages can both recognize H. capsulatum, different receptors involved in recognition can lead to different fates for the fungus43. Macrophages recognize H. capsulatum heat-shock protein 60 by the CD18 integrin, which leads to uptake into an environment in which the fungus is able to live and replicate43. In contrast, DCs are able to kill H. capsulatum and even though they possess the CD18 integrin on their surface, DCs recognize cyclophiln A on H. capsulatum using the fibronectin receptor VLA-543. DC killing of H. capsulatum is due to phagolysosomal fusion and DC lysosomal hydrolases19,43. Human DCs are also able to phagocytose, kill and process Candida albicans for antigen presentation18. The DCs were able to kill Candida as efficiently as macrophages through nonoxidative mechanisms18. An understanding of the mechanisms of DC lysosomal killing is an important area of research due to the ability of Cryptococcus to live and replicate inside macrophages during a pulmonary infection44.
DCs must be able to kill, process and present antigen to initiate the adaptive immune response. Thus, it is important to understand how DCs are able to kill C. neoformans and other fungi. DCs have been shown to phagocytose C. neoformans, where it can be degraded in the lysosomal compartment and processed for antigen presentation20. They have been shown to kill C. neoformans by both oxidative and non-oxidative mechanisms23. Our current studies focused on the non-oxidative mechanisms, such as the lysosomal cathepsin enzymes. We chose to focus on the cathepsins for these studies because they are known lysosomal enzymes that are important in degradation of antigens destined for presentation by DCs. Due to the role of DCs as antigen presenting cells (APCs), it is reasonable that killing the phagocytosed organism would be advantageous, as it would facilitate antigen presentation. Furthermore, due to cathepsin B's role in digesting peptides for MHC class II antigen presentation and our findings that cathepsin B has anti-fungal activity against C. neoformans strain H99, it is likely that this enzyme has dual roles in both killing and in antigen presentation in DCs following phagocytosis of the organism.
While our data show that cathepsin B can inhibit the growth of C. neoformans, we recognize that there are other lysosomal components likely participating in the killing of C. neoformans, and cathepsin B does not act alone. Current studies are focused on fractionation of DC lysosomal extracts to identify additional lysosomal components involved in antifungal activity. We also have preliminary data to suggest that DC lysosomal extract has antifungal activity against other pathogenic fungi including Candida albicans and Coccidioides immitis (unpublished observations) and we will continue to explore the mechanisms of antifungal activity. In this study, we found that the purified lysosomal cathepsin B has inhibitory effects on the growth of C. neoformans. The inhibition is concentration-dependent and we have demonstrated that cathepsin B is found in our lysosomal extracts. Inhibition of purified cathepsin B activity with the three different inhibitors resulted in enhanced activity. While the inhibitors varied in their specificity for cathepsin B, using general cathepsin inhibitors was not an issue due to the fact that we only used purified cathepsin B for our assays. Our data suggest that DC lysosomal cathepsins play a role in the antifungal activity of lysosomal extracts against C. neoformans and interestingly, the inhibition of the enzymatic activity of cathepsin B resulted in enhanced antifungal activity. Currently, we do not know the mechanism of this enhanced effect and studies are underway to explore this phenomenon.
One possible mechanism for the action of cathepsin B with its inhibitors in killing Cryptococcus is that they may act as antimicrobial peptides, due to the cell wall ruptures observed by TEM (Figure 5). Antimicrobial peptides (AMPs) are part of the innate immune system and are found primarily in the skin and the mucosal surfaces. They can also be found in certain immune cells such as neutrophils. The majority of AMPs work by causing a disruption in the cell wall of the organism, which correlates with our data presented in these studies. They accomplish this by forming pores in the cell wall or by coating the organism and when a critical density is reached this causes significant curvature strain that ultimately leads to cell wall rupture (reviewed in33). AMPs have been shown to be important in the host defense against bacteria, viruses, parasites and other fungi34,35. There has been increased interest in AMPs recently in developing them into antimicrobial drugs. Due to the fact that AMPs are able to kill multi-drug-resistant organisms, they are potential antimicrobial drug compounds33,35,45. Treatment of more than 70 clinical isolates of pathogenic fungi, including several drug-resistant Candida and Cryptococcal strains, with five structurally different cathelicidin AMPs, lead to complete killing of the isolates45. There are three main classes of antimicrobial peptides in mammals, including α-defensins, β-defensins and the cathelicidins35. The defensins are cysteine rich and the cathelicidins have a cathelin-like domain which has an amino-terminal cathepsin L inhibitor sequence33. Based on the cell wall rupture induced by cryptococcal treatment with cathepsin B and calpain inhibitor, as seen in the TEM images in figures 5B and 6C, we propose that these components may act as antimicrobial peptides against C. neoformans. The cell wall ruptures induced by cathepsin B and inhibitor treatment caused osmotic lysis of the cryptococcal cells, resulting in cryptococcal death. However, by plating these cells on osmotically supportive agar, we abrogated the killing of C. neoformans.
In conclusion, we showed that DC lysosomal extract has antifungal activity against strains representing all four serotypes of Cryptococcus. In addition, lysosomal cathepsins can inhibit growth of C. neoformans strain H99 and inhibition of the enzymatic activity of cathepsin B leads to enhanced killing of the organism. We demonstrated that treatment with cathepsin B and its inhibitors lead to structural changes and rupture of the fungal cell wall, similar to activity by antimicrobial peptides and we found that the cell death was due to osmotic lysis of the fungus. These findings suggest that we may be able to develop novel antifungal treatments by harnessing the antimicrobial activity of DC lysosomal components.
Methods
Reagents
Unless otherwise stated, chemical reagents were obtained from Sigma-Aldrich (St. Louis, MO), tissue culture media was purchased from Invitrogen (Grand Island, NY) and plasticware was purchased from Fisher Scientific (Pittsburgh, PA). Media for bone marrow-derived dendritic cell (BMDC) generation consisted of RPMI 1640 supplemented with 10% heat-inactivated fetal bovine serum, 2 mM L-glutamine, 100 U penicillin/ml, 100 µg of streptomycin/ml and 50 mM 2-mercaptoethanol (complete medium).
Mice
Female BALB/c (H-2d) mice were purchased from the National Cancer Institute/Charles River Laboratories and were housed at The University of Texas at San Antonio Small Animal Laboratory Vivarium. Mice were handled according to approved guidelines and the study and experimental design was approved by The University of Texas at San Antonio Institutional Animal Care and Use Committee (IACUC).
Culture of Cryptococcus
Cryptococcus neoformans serotype A strain H99, Cryptococcus gattii serotype B strain R265 (gift of Joseph Heitman, Duke University Medical Center, Durham, NC), C. gattii serotype C strain WSA87 (gift of Brian Wickes, University of Texas Health Science Center; San Antonio, TX) and C. neoformans serotype D strain R4249 were recovered from 15% glycerol stocks stored at –80°C and maintained on a yeast-extract-peptone-dextrose (YPD) media (Difco). Yeast cells were grown for 18 hours at 30°C with shaking in YPD broth. Cells were centrifuged and washed three times with sterile phosphate-buffered saline (PBS). Viable yeasts were quantified using trypan blue dye exclusion in a hemacytometer.
BMDCs
BMDC culture was performed as previously described20,23,46. Briefly, bone marrow was flushed from the femurs and tibiae of mice. Cells were washed, counted and plated at a concentration of 2 x 105 cells/ml in complete medium supplemented with 20 ng/ml recombinant murine GM-CSF (Peprotech, Rocky Hill, NJ). Cells were incubated at 37°C, 5% CO2. Half of the medium was replaced every three days and cells were harvested on day 8 following plating. DCs were purified by positive selection using magnetically labeled CD11c antibodies according to the manufacturer's protocol (Miltenyi Biotec, Auburn, CA).
Generation of crude lysosomal extract from BMDCs
Crude BMDC lysosomal extracts were obtained as previously described20 using the lysosome isolation kit (Sigma-Aldrich, St. Louis, MO). Briefly, lysed DCs were centrifuged at 1000 x g for 10 minutes to remove intact cells and cellular debris. The first supernatant was removed and centrifuged at 20,000 x g for 20 minutes to pellet lysosomes. The pellet containing lysosomes was resuspended and sonicated for 20 seconds at 40% power on a Model 300 VT Ultrasonic Homogenizer (BioLogics, Inc., Manassas, VA), resulting in crude lysosome extract (600 µg/ml).
In vitro examination of cryptococci in BMDCs
Purified BMDCs were incubated with C. neoformans yeast cells at a 2:1 ratio with 1 µg/ml of Oregon green-labeled 3C2 opsonizing antibody (gift of Thomas Kozel, University of Nevada, Reno, NV) and 50 nM LysoTracker red (Molecular Probes). Samples were incubated at 37°C for 1 hour prior to imaging. Confocal imaging was performed on a Leica TCS SP2 inverted confocal microscope (Leica Microsystems, Wetzlar, Germany) and Leica confocal software (LCS) was used for the acquisition of images. The images were viewed using a 63X, 1.40-numerical-aperture oil-immersion Leica lens and were digitally increased in size fourfold by using LCS.
Incubation of lysosomal extracts with Cryptococcus
Cryptococcal killing assays were performed as previously described20. Strains were resuspended in 10 mM phosphate buffer with 2% RPMI, pH 5.5 (phosphate buffer). Fungi were added to a 96-well plate in a volume of 50 µl (2.5×105/ml). Phosphate buffer was added to control wells and 50 µl of lysosomal extract20 was added to test wells, for a total volume of 100 µl/well in duplicate wells. Plates were incubated for 24 hours at 37°C, 5% CO2. Cryptococci were diluted in sterile PBS and plated on YPD agar to determine colony forming units (CFU). Killing was defined as CFU significantly below the inoculum and inhibition was defined as CFU significantly below that of growth in phosphate buffer alone.
Incubation of purified lysosomal enzymes with Cryptococcus
Purified cathepsins B, D and L (Enzo Life Sciences, Inc., Farmingdale, NY) at 10 μg/ml (physiological concentration)38,39 and 50 μg/ml (excess concentration) were incubated with C. neoformans as previously described for lysosomal extract experiments. All enzymes were commercially manufactured and are >95% pure. Plates were incubated for 24 hours at 37°C, 5% CO2. Following incubation, organisms were plated for CFU.
Cathepsin B Western blot
For Western blotting, 15 μl of the lysosomal extracts (10 µg protein) or 15 µl (1500 pg) of recombinant mouse cathepsin B (R & D Systems, Minneapolis, MN) were mixed with 5 μl of 4X Laemmli buffer and boiled for 5 minutes. Recombinant mouse cathepsin B in the pro-form (39 kDa) was used as a positive control. Active cathepsin B is approximately 25–26 kDa. For SDS-PAGE, 5 µl of Precision Plus Protein Dual Color Standards (BIO-RAD) and 20 µl of each lysosomal extract and purified cathepsin B (positive control) were loaded into a 10% Mini-PROTEAN TGX gel (BIO-RAD). The gel was run for 40 minutes at 200 V, transferred onto a PVDF membrane (Thermo Scientific, Rockford, IL) and blocked with 5% skim milk in Tris-buffered saline with 1% Tween 20 (TBS-T) at room temperature. After 1 h, the membrane was incubated with 1 µg/ml anti-human/mouse cathepsin B (R & D Systems) in 5% milk/TBS-T for 2 hours at room temperature. The membrane was washed three times for 5 min with TBS-T and then incubated with anti-rat IgG HRP (R & D Systems) at a 1:1000 dilution in 5% milk/TBS-T for 1 h at room temperature. The membrane was washed three times for 5 min with TBS-T and then two times for 10 min with TBS followed by incubation with SuperSignal West Dura Extended Duration substrate (Thermo Scientific) for 5 minutes. The membrane was exposed to CL-XPosure Film (Thermo Scientific) and developed using an AFP Imaging Mini-Medical series developer (Medlink Imaging Teterboro, NJ).
Cathepsin B ELISA
Total cathepsin B in lysosomal extracts was quantified by ELISA. The capture and detection antibodies and recombinant mouse cathepsin B (used for the standard) were purchased from R & D Systems (Minneapolis, MN). Microtiter plates were coated with 1 µg/ml of anti-human/mouse cathepsin B diluted in PBS and incubated overnight at 4°C. After washing three times with PBS-Tween buffer (1X PBS and 0.05% Tween 20), the wells were blocked with 10% goat serum in 1X PBS at room temperature for 1 h. After blocking, the plates were washed three times and the samples (diluted 1:1000) and the standards were added. After 2 h incubation at room temperature, the plates were washed three times. Then, biotinylated anti-mouse cathepsin B diluted to 1 µg/mL in reagent diluent (1% goat serum in 1X PBS) was added. After incubation for 1.5 h at room temperature, the plates were washed and streptavidin-HRP (R & D Systems) was added. After 25 minutes, the plates were washed 5 times and 1X TMB substrate (eBioscience, San Diego, CA) was added and incubated in the dark for 15–20 minutes. The reaction was stopped by adding 2N H2SO4. Samples were read at 405 nm on a BioTek EL808 automated plate-reader (BioTek, Winooski, VT) and analyzed with Gen 5 software (BioTek).
Cathepsin B inhibition
Calpain inhibitor (Sigma-Aldrich Co), cathepsin B & L inhibitor (BioVision, Inc., Mountain View, CA), or CA-074 (Enzo Life Sciences) was added to C. neoformans at 10 µg/ml, 50 or 100 µM, or 20 µM, respectively and incubated with or without purified cathepsin B at 50 μg/ml, following the same procedure indicated above. Following 24 h incubation, organisms from wells were diluted in sterile PBS and plated on YPD agar to determine CFU/ml. For osmotic stress experiments, cathepsin B and calpain inhibitor were incubated with C. neoformans strain H99 for 24 hours at 37°C, 5% CO2 and then either plated on YPD agar or on osmotically supportive YPD containing 1.4 M sorbitol. Percent cathepsin B enzymatic activity was measured using the Cathepsin B Activity Assay Kit (BioVision, Inc., Mountain View, California) according to manufacturer's specifications and read with a Synergy HT fluorescent plate reader (BioTek) at an excitation of 400-nm and an emission of 505-nm and analyzed with Gen 5 software (BioTek).
Electron microscopy
Cryptococcus neoformans strain H99 (serotype A) was incubated with cathepsin B and/or inhibitors as described above for 12 hours. For osmotic stress EM experiments, cells were incubated for 24 h then transferred to YPD or YPD + 1.4 M sorbitol broth for 8 h at 30°C. Cells were fixed with 4% formaldehyde/1% glutaraldehyde and processed for scanning electron microscopy (SEM) or transmission electron microscopy (TEM) by the electron microscopy staff in the Pathology department at the University of Texas Health Science Center San Antonio. Following sample preparation, cells were examined on a JSM-6610 SEM or on a Philips 208 S TEM. Images were examined at 2500X and 5000X for SEM and 25000X and 4000X for TEM.
Statistical analysis
One-way ANOVA with the Tukey's multiple comparison test was used to compare CFU using GraphPad Prism version 5.00 for Windows (GraphPad Software, San Diego, CA). Significant differences were defined as p < 0.05.
References
Levitz, S. M. The ecology of Cryptococcus neoformans and the epidemiology of cryptococcosis. Rev Infect Dis 13, 1163–1169 (1991).
Mitchell, T. G. & Perfect, J. R. Cryptococcosis in the Era of AIDS - 100 years after the discovery of Cryptococcus neoformans. Clin Microbiol Rev 8, 515–548 (1995).
Park, B. J. et al. Estimation of the current global burden of cryptococcal meningitis among persons living with HIV/AIDS. AIDS 23, 525–530 (2009).
Pappas, P. G. et al. Invasive fungal infections among organ transplant recipients: results of the Transplant-Associated Infection Surveillance Network (TRANSNET). Clin Infect Dis 50, 1101–1111, 10.1086/651262 (2010).
Powderly, W. G. Cryptococcal Meningitis and AIDS. Clinical Infectious Diseases 17, 837–842 (1993).
Huffnagle, G. B., Yates, J. L. & Lipscomb, M. F. T-cell-mediated immunity in the lung - a Cryptococcus neoformans pulmonary infection model using SCID and athymic nude-mice. Infect Immun 59, 1423–1433 (1991).
Hill, J. O. & Harmsen, A. G. Intrapulmonary growth and dissemination of an avirulent strain of Cryptococcus neoformans in mice depleted of CD4+ or CD8+ T-Cells. J Exp Med 173, 755–758 (1991).
Huffnagle, G. B., Lipscomb, M. F., Lovchik, J. A., Hoag, K. A. & Street, N. E. The role of CD4(+) and CD8(+) T-Cells in the protective inflammatory response to a pulmonary cryptococcal infection. J Leukoc Biol 55, 35–42 (1994).
Mody, C. H., Lipscomb, M. F., Street, N. E. & Toews, G. B. Depletion of CD4+ (L3T4+) lymphocytes in vivo impairs murine host defense to Cryptococcus neoformans. J Immunol 144, 1472–1477 (1990).
Murphy, J. W. Protective cell-mediated immunity against Cryptococcus neoformans. Res Immunol 149, 373–386 (1998).
Wozniak, K. L. et al. Insights into the mechanisms of protective immunity against Cryptococcus neoformans infection using a mouse model of pulmonary cryptococcosis. PLoS One 4 (2009).
Steinman, R. M. Dendritic cells and the control of immunity: enhancing the efficiency of antigen presentation. Mt Sinai J Med 68, 160–166 (2001).
Kubach, J. et al. Dendritic cells: sentinels of immunity and tolerance. Int J Hematol 81, 197–203 (2005).
Iwasaki, A. Mucosal dendritic cells. Annu Rev Immunol 25, 381–418 (2007).
Banchereau, J. et al. Immunobiology of dendritic cells. Annu Rev Immunol 18, 767–811 (2000).
Mellman, I. Antigen processing and presentation by dendritic cells: Cell biological mechanisms. Adv Exp Med Biol 560, 63–67 (2005).
Mambula, S. S., Simons, E. R., Hastey, R., Selsted, M. E. & Levitz, S. M. Human neutrophil-mediated nonoxidative antifungal activity against Cryptococcus neoformans. Infect Immun 68, 6257–6264 (2000).
Newman, S. L. & Holly, A. Candida albicans is phagocytosed, killed and processed for antigen presentation by human dendritic cells. Infect Immun 69 (2001).
Gildea, L. A., Ciraolo, G. M., Morris, R. E. & Newman, S. L. Human dendritic cell activity against Histoplasma capsulatum is mediated via phagolysosomal fusion. Infect Immun 73, 6803–6811 (2005).
Wozniak, K. L. & Levitz, S. M. Cryptococcus neoformans enters the endolysosomal pathway of dendritic cells and is killed by lysosomal components. Infect Immun 76, 4764–4771 (2008).
Bozza, S. et al. Dendritic cells transport conidia and hyphae of Aspergillus fumigatus from the airways to the draining lymph nodes and initiate disparate Th responses to the fungus. J Immunol 168, 1362–1371 (2002).
LeBlanc, D. M., Barousse, M. M. & Fidel, P. L., Jr Role for Dendritic Cells in Immunoregulation during Experimental Vaginal Candidiasis. Infect. Immun. 74, 3213–3221 (2006).
Kelly, R. M., Chen, J. M., Yauch, L. E. & Levitz, S. M. Opsonic requirements for dendritic cell-mediated responses to Cryptococcus neoformans. Infect Immun 73, 592–598 (2005).
Wozniak, K. L., Vyas, J. M. & Levitz, S. M. In Vivo Role of Dendritic Cells in a Murine Model of Pulmonary Cryptococcosis. Infect. Immun. 74, 3817–3824 (2006).
Colbert, J. D., Matthews, S. P., Miller, G. & Watts, C. Diverse regulatory roles for lysosomal proteases in the immune response. Eur J Immunol 39, 2955–2965 (2009).
Honey, K. & Rudensky, A. Y. Lysosomal cysteine proteases regulate antigen presentation. Nat Rev Immunol 3, 472–482 (2003).
Zavasnik-Bergant, T. & Turk, B. Cysteine cathepsins in the immune response. Tissue Antigens 67, 349–355 (2006).
Riese, R. J. & Chapman, H. A. Cathepsins and compartmentalization in antigen presentation. Curr Opin Immunol 12, 107–113 (2000).
Lennon-Dumenil, A. M. et al. Analysis of protease activity in live antigen-presenting cells shows regulation of the phagosomal proteolytic contents during dendritic cell activation. J Exp Med 196, 529–540 (2002).
Savina, A. & Amigorena, S. Phagocytosis and antigen presentation in dendritic cells. Immunol Rev 219, 143–156 (2007).
Illy, C. et al. Role of the occluding loop in cathepsin B activity. J Biol Chem 272, 1197–1202 (1997).
Guncar, G. et al. Crystal structure of porcine cathepsin H determined at 2.1 A resolution: location of the mini-chain C-terminal carboxyl group defines cathepsin H aminopeptidase function. Structure 6, 51–61 (1998).
Giuliani, A., Pirri, G. & Nicoletto, S. Antimicrobial peptides: an overview of a promising class of therapeutics. Central European Journal of Biology 2, 1–33 (2007).
Fjell, C. D., Hiss, J. A., Hancock, R. E. & Schneider, G. Designing antimicrobial peptides: form follows function. Nat Rev Drug Discov 11, 37–51 (2012).
Peters, B. M., Shirtliff, M. E. & Jabra-Rizk, M. A. Antimicrobial peptides: primeval molecules or future drugs? PLoS Pathog 6 (2010).
Trombetta, E. S., Ebersold, M., Garrett, W., Pypaert, M. & Mellman, I. Activation of lysosomal function during dendritic cell maturation. Science 299, 1400–1403 (2003).
Trombetta, E. S. & Mellman, I. Cell biology of antigen processing in vitro and in vivo. Annu Rev Immunol 23, 975–1028 (2005).
Premzl, A., Turk, V. & Kos, J. Intracellular proteolytic activity of cathepsin B is associated with capillary-like tube formation by endothelial cells in vitro. J Cell Biochem 97, 1230–1240 (2006).
Werle, B., Julke, B., Lah, T., Spiess, E. & Ebert, W. Cathepsin B fraction active at physiological pH of 7.5 is of prognostic significance in squamous cell carcinoma of human lung. Br J Cancer 75, 1137–1143 (1997).
Tanaka, Y., Tanaka, R., Kawabata, T., Noguchi, Y. & Himeno, M. Lysosomal cysteine protease, cathepsin B, is targeted to lysosomes by the mannose 6-phosphate-independent pathway in rat hepatocytes: site-specific phosphorylation in oligosaccharides of the proregion. J Biochem 128, 39–48 (2000).
Ibrahim-Granet, O. et al. Phagocytosis and intracellular fate of Aspergillus fumigatus conidia in alveolar macrophages. Infect Immun 71, 891–903 (2003).
Nicola, A. M. & Casadevall, A. In vitro measurement of phagocytosis and killing of Cryptococcus neoformans by macrophages. Methods Mol Biol 844, 189–197 (2012).
Gomez, F. J., Pilcher-Roberts, R., Alborzi, A. & Newman, S. L. Histoplasma capsulatum cyclophilin A mediates attachment to dendritic cell VLA-5. J Immunol 181, 7106–7114 (2008).
Feldmesser, M., Kress, Y., Novikoff, P. & Casadevall, A. Cryptococcus neoformans is a facultative intracellular pathogen in murine pulmonary infection. Infect Immun 68, 4225–4237 (2000).
Benincasa, M. et al. Fungicidal activity of five cathelicidin peptides against clinically isolated yeasts. J Antimicrob Chemother 58, 950–959 (2006).
Lutz, M. B. et al. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods 223, 77–92 (1999).
Acknowledgements
The authors wish to thank Stuart M. Levitz for his helpful comments and for critical reading of this manuscript. The authors also wish to thank Barbara Hunter and the staff of the UTHSCSA electron microscopy facility for their assistance with the electron microscopy studies. This work was supported by grant RO1 AI071752-04 from the National Institute of Allergy and Infectious Diseases (NIAID) of the National Institutes of Health (NIH) (F.L.W.).
Author information
Authors and Affiliations
Contributions
CRH and KLW wrote the main manuscript text, CRH, HB and KLW prepared all figures, CRH, HB and KLW conducted the experiments. HB, CRH, KLW and FLW analyzed the data. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Hole, C., Bui, H., Wormley, F. et al. Mechanisms of Dendritic Cell Lysosomal Killing of Cryptococcus. Sci Rep 2, 739 (2012). https://doi.org/10.1038/srep00739
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep00739
This article is cited by
-
Antifungal activity of dendritic cell lysosomal proteins against Cryptococcus neoformans
Scientific Reports (2021)
-
Role of Cathepsins in Mycobacterium tuberculosis Survival in Human Macrophages
Scientific Reports (2016)
-
Innate host defenses against Cryptococcus neoformans
Journal of Microbiology (2016)
-
Dancing cheek to cheek: Cryptococcus neoformans and phagocytes
SpringerPlus (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
