
Abstract
Although amorphous structures have been widely obtained in various multi-component metallic alloys, amorphization in pure metals has seldom been observed and remains a long-standing scientific curiosity and technological interest. Here we present experimental evidence of localized solid-state amorphization in bulk nanocrystalline nickel introduced by quasi-static compression at room temperature. High-resolution electron microscope observations illustrate that nano-scale amorphous structures present at the regions where severe deformation occurred, e.g. along crack paths or surrounding nano-voids. These findings have indicated that nanocrystalline structures are highly desirable for promoting solid-state amorphization, which may provide new insights for understanding the nature of the crystalline-to-amorphous transformation and suggested a potential method to produce elemental metallic glasses that have hardly been available hitherto through rapid solidification.
Similar content being viewed by others

Introduction
Producing non-crystalline (or amorphous) structures in metallic materials has long been a focus for vast amount of theoretical and experimental investigations in materials science1,2,3,4,5,6. There have been two routes for achieving amorphous structures in metallic materials: (i) “freezing” the dynamic disorder of liquid under extremely high cooling rate, i.e. rapid quenching7; (ii) destabilizing and “melting” a solid to amorphous state by introducing chemical and/or structural disorder, which is known as solid-state amorphization (SSA)8. In both cases, however, almost all the amorphous metallic materials so far produced are complex alloys containing two or more constituents9. Although making elemental metals amorphous is important both scientifically, for comprehensive understanding the glass formation process10 and technologically, for eliminating phase separation which is detrimental to the thermal and mechanical properties of metallic glasses (MGs)11, successes have been demonstrated for only a few cases and all of them necessitate extreme experimental conditions (e.g. elevated temperature combined with ultrahigh static pressure9 or application of stringent kinetic constraints12). Molecular dynamic (MD) simulations predict that single crystal Ni nanowires could be transformed into an amorphous state, but only when they deformed under an extremely high strain rate13. In this sense, obtaining amorphous structures in pure metals, especially through conventional processes under normal conditions, is yet a remaining challenge.
Recent studies regarding size-dependent thermodynamic properties of nanocrystalline (NC) solids suggested that some materials that have never been made amorphous can become highly susceptible to irradiation-induced amorphization when they are in the form of nano-scale precipitates embedded in an amorphous matrix14. It is supposed that the drastic increasing of surface energy due to particle-size refinement may potentially lower the energy barrier, which subsequently promotes the crystalline-to-amorphous (c-a) transformation14. This implies that transforming a crystalline material with a high-energy state to an amorphous phase might be easy since the extra initial free energy can contribute significantly to the thermodynamic driving force for the c-a transformation. The same strategy can also be applied to amorphizing a elemental metallic system by using bulk NC metals directly as starting materials: The relatively high energy level coming from the significant volume fraction of structurally disordered grain boundaries (GBs) in NC materials may favor to destabilize the crystalline structure15, making amorphization more prone to occur, even merely through ordinary solid-state processes. However, to the best of our knowledge, no relevant experimental evidence has been available.
In this study we show that by employing NC Ni as the precursor, localized SSA can be achieved simply through conventional mechanical loading (room-temperature quasi-static compression). The amorphous structures can, in turn, influence the deformation and fracture behaviors of the NC Ni.
Results
Materials and compression tests
The NC Ni used in this study was synthesized by electrodeposition. Transmission electron microscopy (TEM) observations (Fig. 1a) indicated that the NC Ni consisted of roughly equiaxed grains with random orientations and the average grain size was estimated to be 20 nm. Most of the nanograins were separated by high-angle GBs whereas no GB phase (e.g. amorphous layers) was detected. A high purity of the as-deposited NC Ni sheets (99.92%, weight percent) was revealed by chemical analysis. Room temperature uniaxial compression tests were performed at a strain rate of 10−5 s−1. Further details on sample preparation and compression test can be found in the Methods section and Ref. 15. As shown by the true stress-strain curve in Figure 1b, during compression the NC Ni exhibits remarkably enhanced plasticity concurrent with ultra-high flow stress: the strain to failure is ~36% and the maximum compressive strength is 2079 MPa. Such large plasticity enables us to systematically study the structural evolution and the underlying deformation mechanism. Scanning electron microscope (SEM) observations on the fractured surface illustrated the widespread distribution of nano-sized voids (Fig. 1c, marked by arrows) and micro-crack networks (Fig. 1d), both of which were formed during deformation and influenced the fracture process16.

Microstructures and compressive behavior of NC Ni.
(a) Bright-field TEM image showing the typical microstructure of as-deposited NC Ni. Inset gives the selected-area electron diffraction pattern. (b) Quasi-static compressive true stress-strain curve for NC Ni sample tested at room temperature. SEM fractography examinations illustrate two representative features on the fractured surface: (c) nano-sized voids (marked by white arrows) and (d) micro-crack networks.
Localized solid-state amorphization
Figure 2a is a typical low-magnification TEM image of a NC Ni sample after deformation, showing a crack-like damage zone with uniform light contrast. Similar zones were identified in many places in the sample. An extensive high-resolution TEM (HRTEM) investigation (see, Fig. 2b, for example) reveals that some parts of the damage zones are not cracks, but rather 3~5 nm wide uncracked bands with no lattice fringes inside. A fast Fourier transform (FFT) pattern taken from the band (Fig. 2c) shows a diffuse halo with no diffraction spots, confirming the amorphous nature of the area. By contrast, the FFT pattern of the crystalline matrix nearby (Fig. 2d) gives obvious evidence of the crystal structure of face-centered cubic (FCC) Ni. The angular mismatch between the lattice fringes on the two sides of the amorphous band is ~1°, revealing that this section of the amorphous band was formed directly inside the grain interior (Fig. 2b). Figure 2e depicts another HRTEM image showing a larger area around the same amorphous band in Figure 2b. Seen from this figure, some deformation debris, such as stacking faults (marked by Sf) and a narrow heavy deformation band (HDB) (delineated by two white dashed lines), are apparently visible. The HDB appears as mesocrystalline region with ill-defined lattice image. A Fourier-filtered image of the area in Fig. 2f clearly illustrates that HDB is in fact a mixture of amorphous phases interweaved with nano-crystals (Fig. 2f). Amorphous layers which manifest themselves as light-contrast regions in low-magnification TEM images (Fig. 3a) and maze-like patterns in HRTEM images (Fig. 3b) were also found surrounding some nano-voids and these layers range in width from 5 to 20 nm. Chemical analysis of the amorphous and crystalline regions conducted using TEM energy disperse spectroscopy (EDS) shows no detectable compositional difference among the regions, implying that the amorphization did not result from introduction of chemical disorder or formation of some oxides. In view of the test conditions (room temperature of ~300 K and quasi-static strain rate of 10−5 s−1) and the fact that no evidence of melting such as flow features and diffuse boundaries was found by TEM and HRTEM observations, the possibility of local melting and re-solidification into amorphous phase can be ruled out. Derived from these observations, we suggest that the observed amorphization occurred as a solid-state process during deformation.

Post-deformation microstructures of NC Ni.
(a) Bright-field TEM image showing linear crack-like damage zones with uniform light contrast. (b) HRETM image of the red-boxed area in (a) revealing a 3~5 nm wide band with free-crystallographic contrast. Misalignment of the lattice images on either side of the band was measured to be ~1°, by extension the fringes across the band (denoted by blue and yellow straight lines). (c) and (d) FFT patterns from inside the band and the crystalline region nearby, respectively. The loss of lattice fringes in the band, combined with the diffuse hallo in the FFT pattern, provide direct evidence of localized amorphization. (e) HRTEM image of the green-boxed area in (a). Some deformation debris can be conspicuously noted. Sf indicates stacking faults and the HBD is highlighted by two white dashed lines. (f) A Fourier-filtered image from inside the white-boxed area in (e) for a close up view of the HDB.

Amorphous structures around nano-voids.
(a) Low-magnification bright-field TEM image of the amorphous structures, which appear as light-contrast layers, around a peanut-shaped nano-sized void. (b) HRTEM image of the white rectangular area in (a), showing modulated contrast typical to a single amorphous structure. FFT pattern [Inset in (b)] displays a diffuse hallo, supporting the amorphous nature of the detected area.
Discussion
A central question to be addressed in our present study is what is responsible for the amorphization of NC Ni under compression. It is evident from the TEM observations that amorphization mostly occurs either along crack paths or surrounding nano-voids, around which the areas experience larger deformation than the global level to accommodate the extra plastic flow during void formation as well as crack propagation. This observation, combined with the finding of extensive deformation imprints around amorphous zones, suggest that plastic deformation should play the dominant role in the observed c-a transformation. Such a deduction can be further corroborated by the finding of highly disordered regions (HDRs) around some nano-voids, as shown in Figure 4a. HRTEM images reveal that such HDRs consist of highly distorted tiny crystalline domains separated by disordered or even amorphous regions (Fig. 4b), which structurally resemble the HDBs (Fig. 2f). These structural features illustrate that crystalline lattice experienced severe disturbance under high degree of localized deformation and the original nano-sized grains were further broken up into even smaller ones with size of 3~5 nm. Grain-refinement was frequently observed in multi-component alloys or intermetallics subject to severe plastic deformation3,17 and was suggested to be the prior step to the onset of amorphization18. The observed grain refinement in the present case can be interpreted in terms of deformation-induced structural disordering. MD simulations predict that in NC materials when the applied strain within a definite region is beyond a critical level, mechanically driven structural disordering can be triggered as an accommodation mechanism to release the extremely high local stress and to mediate the plasticity19. Structural disordering was also supposed to be the dominant deformation mechanism in some nano-dimensional materials under large strains, such as SiC nanowires20,21. The disordering process, in essence, contributes the grain refinement in two aspects. On one hand, considering the non-equilibrium nature of the GB regions in NC metals22, disordering may first initiate in GBs then spread into grain interiors, leading to decreasing in grain size; on the other hand, those newly formed intra-grain disordered/amorphous regions can be thought of as GBs inducing a refining effect by subdividing the grains into smaller domains. From the thermodynamic point of view, reducing grain size is tantamount to introducing more defects (GBs), giving rise to an increase in the stored energy of a system. As noted in the introduction, the original free energy of NC metals is on a relatively high level. Therefore the defect accumulation due to plastic deformation can, by starting off higher on the free energy “ladder”, raise an elemental metallic-bond system to a non-equilibrium state energetically above the amorphous state, such that the energy difference between these two states drives the amorphization23. Another factor should be taken into consideration is that relative to the interface between two crystallites, the crystalline-amorphous interface has a smaller energy, which favors the formation of amorphous phase24. In the kinetic perspective, the aforementioned disordering proceeds by displacing atoms away from their equilibrium positions, which can be realized through local shear displacement25, atomic shuffling and stress-assisted free-volume migration26. As disordered regions continue to nucleate, expand and intersect with each other, whenever the mean displacement of most atoms within a local region approaches a critical value therefore they are unable to arrange themselves according to the original crystalline structure, amorphization takes place25. The structural details of HDRs and HDBs reveal the existence of an intermediate stage bridging the crystalline and amorphous phases during amorphization. The c-a transformation may originate within HDRs and HDBs then extend. Accordingly bearing in mind the structural similarity between HDR and HDB, it appears that the above-delineated amorphization mechanism operated during the formation of both amorphous bands along crack paths and amorphous layers around nano-voids.

TEM and HRTEM observations of the highly disordered regions around nano-voids.
(a) Low-magnification bright-field TEM image. (b) HRTEM image of the part framed by white rectangular box in (a). Circles in (b) indicate discernible nano-crystals surrounded by amorphous phases.
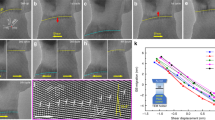
Further structural examinations reveal that there are other defects, besides GBs, contribute to the observed c-a transformation. Fig. 5a depicts another example of an amorphous band. Clearly visible on the periphery of this band are a large number of trapped lattice dislocations (marked with red “T” symbols), a signature of extensive localized plastic deformation, whereas no clear evidence of grain refinement, such as the presence of HDB or HDR, was found. Careful analysis reveals that the typical dislocation structures in the deformed NC Ni are a/2 <110> full dislocations. The enlarged Fourier-filtered image of one representative dislocation is given in Fig. 5c for a better perspective of details and by drawing local Burgers circuit its Burgers vector is identified to be a/2 [−101] or a/2 [110], which has an angle of 60° (or 120°) with respect to the dislocation line. Such observations align well with the previously reported results which frequently demonstrated the existence of trapped dislocations in deformed NC Ni with grain size even as small as 10 nm27,28. In NC metals dislocations generate from GB sources (stress concentrations at GBs29 or bow-out of pre-existing GB dislocations30) to sustain plasticity and to accommodate processes such as GB sliding and grain rotation26. Particularly, due to the limited spaces offered by nano-sized grains and strong encumbrance for dislocation movements stemming from copious interfaces such as twin-boundaries or GBs, dislocations from multiple sources always concentrate to a small deformation volume31,32, leading to extremely high local dislocation density. From the statistical analysis of the one-dimensional Fourier-filter image of the selected area in the vicinity of the amorphous band (see, Fig. 5d , for example), the dislocation densities on (111) and (−111) crystallography planes are estimated to be 7.6×1016/m2 and 17.9×1016/m2, respectively. So the total dislocation density within this local region is as high as around 25.5×1016/m2, which is comparable with the previously reported values that can directly induce SSA17,32,33. The exceptionally high dislocation density in a local region engenders lattice distortion which might be severe enough to directly initiate SSA34. MD simulations and theoretical models also demonstrated that the dislocation accumulation contributes significantly to the atomic disorder and lattice strain, leading to elastic softening and the final collapse of the crystalline structure35,36,37. In addition, as we discussed above, with the energy increment supplied by dislocation accumulations, the free energy of the original NC structures is more prone to be raised up to a point higher than that of an amorphous phase, providing the thermodynamic driving force for the SSA. Thus a reasonable conclusion can be drawn that dislocations also play a crucial role during the formation of the amorphous structures. Taken as a whole, high densities of GBs and dislocations, both of which are introduced by localized heavy plastic deformation, contribute significantly to destabilizing the crystalline structures and to driving the SSA. Upon such a scenario, nanocrystalline structures facilitate the c-a transformation both thermodynamically and kinetically. One point worthwhile to mention is that the grain-refinement-induced and dislocation-accumulation-induced amorphization operate concomitantly throughout the sample, but it is possible that, for a given region in the sample, only one of the two mechanisms dominates the c-a transformation, probably depending on the local stress state and strain condition.

Dislocation structures around an amorphous band.
(a) HRTEM image showing a high density of dislocations in the area around an amorphous band. (b) the FFT pattern obtained from the area marked with the white square in (a), confirming that the crystalline phase is oriented close to a <110> zone axis. (c) Enlarged Fourier-filtered image of the area marked by white square in (a), revealing the details of a a/2 <110> full dislocation. Local Burgers circuit, starting at S and ending at F, encloses the core part of the dislocation which marked by a blue “T” symbol. (d) A one-dimensional Fourier-filtered image taken near the amorphous band. Dislocations, marked with red “T” symbols, can be clearly seen from the extra (111) half-planes.
Once formed, the amorphous structures exhibit certain level of stability as revealed by the fact that they can be extensively observed in the deformed Ni samples. The question of what gives rise to this stability is not well known at present, but either the constraints coming from surrounding crystalline matrix or the stress field along the crystalline-amorphous interfaces38, we speculate, may help to stabilize the amorphous structures. It is worthwhile to point out that comparing with the as-deposited grain size of ~20 nm, which is quite close to the so-called “strongest size” defined both theoretically39 and experimentally40, the size of the nano-crystals within HDRs and HDBs has fallen into the “inverse Hall-Petch” regime, which means that these regions are much softer than the crystalline parts41. MD simulation also predicted that formation of amorphous phase would cause local weakening42. Hence the soft disordered and amorphous areas can serve as preferential sites to be continuously deformed until void formation or provide easy cleavage paths for crack propagation43. So it is reasonable to conjecture that the amorphous/disordered structures are precursors for some of the observed nano-voids as well as micro-crack networks (Fig. 1c and d) and the amorphization-induced failure is at least in part responsible for the fracture of NC Ni.
Methods
Sample preparation and compression test
The NC Ni samples were produced via surfactant-assisted direct current electrodeposition from a modified Watt's bath mainly containing nickel sulfate (NiSO4), nickel chloride (NiCl2), boric acid (H3BO3) and several kinds of additives. During deposition, the electrolyte was maintained at a pH level of 5.0 and a temperature of 50°C. All operating parameters were carefully controlled to ensure a homogeneous structure and uniform grain size distribution. After deposition, the NC Ni sheets with a thickness of about 6 mm were mechanically stripped from the substrates.
The cylindrical-form samples were machined from the as-deposited sheets by a wire electro discharging machine to a diameter of 3 mm and a height of 4 mm for compression test. All the samples were carefully polished to mirror finish on outside surfaces to eliminate the visible tapering of sidewalls for accurate measurement of the flow stress during compression. Uniaxial compression tests were performed at a strain rate of 10−5 s−1 using MTS 810 system at room temperature with loading along the deposition (thickness) direction.
Characterization techniques
TEM and HRTEM observations were performed using a JEM-2100F with an applied voltage of 200 kV and a point-to-point resolution of ~0.2 nm. The samples for TEM observations were first thinned manually then made electron-transparent by ion-milling using a Leica RES101 Ion Polishing System. SEM fracture morphology observation was done using a ZEISS SUPRA 40. The chemical composition analysis was carried out on inductively coupled plasma atomic emission spectrometry (ICP-AES, Plasma/1000) and carbon/sulfur determinator (CS-200).
References
Klement, W., Willens, R. H. & Duwez, P. Non-crystalline structure in solidified gold–silicon alloys. Nature 187, 869–870 (1960).
He, Y., Poon, S. J. & Shiflet, G. J. Synthesis and properties of metallic glasses that contain aluminum. Science 241, 1640–1642 (1988).
Jang, J. S. C. & Koch, C. C. Amorphization and disordering of the Ni3Al ordered intermetallic by mechanical milling. J. Mater. Res. 5, 498–510 (1990).
Winters, R. R. & Hammack, W. S. Pressure-induced amorphization of R-Al5Li3Cu: a structural relation among amorphous metals, quasi-crystals and curved space. Science 260, 202–204 (1993).
Inoue, A. Stabilization of metallic supercooled liquid and bulk amorphous alloys. Acta Mater. 48, 279–306 (2000).
Hofmann, D. C. et al. Designing metallic glass matrix composites with high toughness and tensile ductility. Nature 451, 1085–1090 (2008).
Greer, A. L. Metallic glasses. Science 267, 1947–1953 (1995).
Fecht, H. J. Defect-induced melting and solid-state amorphization. Nature 356, 133–135 (1992).
Bhat, M. H. et al. Vitrification of a monatomic metallic liquid. Nature 448, 787–791 (2007).
Hoang, V. V. & Odagaki, T. Glass formation and thermodynamics of supercooled monatomic liquids. J. Phys. Chem. B 115, 6946–6956 (2011).
KÜndig, A. A., LÖffler, J. F., Johnson, W. L., Uggowitzer, P. J. & Thiyagarajan, P. Influence of decomposition on the thermal stability of undercooled Zr-Ti-Cu-Ni-Al alloys. Scripta Mater. 44, 1269–1273 (2001).
Holz, M., Ziemann, P. & Buckel, W. Direct evidence for amorphization of pure gallium by low-temperature ion irradiation. Phys. Rev. Lett. 51, 1584–1587 (1983).
Ikeda, H. et al. Strain rate induced amorphization in metallic nanowires. Phys. Rev. Lett. 82, 2900–2903 (1999).
Meldrum, A., Boatner, L. A. & Ewing, R. C. Nanocrystalline zirconia can be amorphized by ion irradiation. Phys. Rev. Lett. 88, 025503 (2002).
Fecht, H. J. Thermodynamic properties of amorphous solids - glass formation and glass transition. Mater. Trans. Japan. Inst. Metals 36, 777–793 (1995).
Shen, X. X., Xu, Z., Lian, J. S. & Jiang, Q. The deformation and fracture behavior of an electrodeposited nanocrystalline Ni under compression. Mater. Sci. Eng. A 528, 7878–7886 (2011).
Huang, J. Y., Zhu, Y. T., Liao, X. Z. & Valiev, R. Z. Amorphization of TiNi induced by high-pressure torsion. Philos. Mag. Lett. 84, 183–190 (2004).
Sagel, A., Wanderka, N., Wunderlich, R. K., Schubert-Bischoff, P. & Fecht, H.-J. Early stages of solid-state amorphization reaction during mechanical alloying of a multicomponent Zr-powder mixture. Scripta Mater. 38, 163–169 (1998).
Szlufarska, I., Nakano, A. & Vashishta, P. A crossover in the mechanical response of nanocrystalline ceramics. Science 309, 911–914 (2005).
Han, X. D. et al. Low-temperature in situ large strain plasticity of ceramic SiC nanowires and its atomic-scale mechanism. Nano Lett. 7, 452–457 (2007).
Zhang, Y. F. et al. Direct observation of super-plasticity of beta-SiC nanowires at low temperature. Adv. Funct. Mater. 17, 3435–3440 (2007).
Gleiter, H. Nanostructured materials: basic concepts and microstructure. Acta Mater. 48, 1–29 (2000).
Fan, G. J., Guo, F. Q., Hu, Z. Q., Quan, M. X. & Lu, K. Amorphization of selenium induced by high-energy ball milling. Phys. Rev. B 55, 11010–11013 (1997).
Benedictus, R., Han, K., Træholt, C., BÖttger, A. & Mittemeijer, E. J. Solid state amorphization in Ni-Ti systems: the effect of structure on the kinetics of interface and grain-boundary amorphization. Acta Mater. 46, 5491–5508 (1998).
Zheng, G.-P. & Li, M. Crystal instability in nanocrystalline materials. Acta Mater. 55, 5464–5472 (2007).
Swygenhoven, H. Van & Derlet, P. M. Grain-boundary sliding in nanocrystalline fcc metals. Phys. Rev. B 64, 224105 (2001).
Shan, Z. W. et al. Dislocation dynamics in nanocrystalline nickel. Phys. Rev. Lett. 98, 095502 (2007).
Wu, X. L., Zhu, Y. T., Wei, Y. G. & Wei, Q. Strong strain hardening in nanocrystalline nickel. Phys. Rev. Lett. 103, 205504 (2009).
Asaro, R. J. & Suresh, S. Mechanistic models for the activation volume and rate sensitivity in metals with nanocrystalline grains and nano-scale twins. Acta Mater. 53, 3369–3382 (2005).
Wang, Y. M., Hamza, A. V. & Ma, E. Activation volume and density of mobile dislocations in plastically deforming nanocrystalline Ni. Appl. Phys. Lett. 86, 241917 (2005).
Wang, Y. B., Liao, X. Z., Zhao, Y. H., Lavernia, E. J., Ringer, S. P., Horita, Z., Langdon, T. G. & Zhu, Y. T. The role of stacking faults and twin boundaries in grain refinement of a Cu-Zn alloy processed by high-pressure torsion. Mater. Sci. Eng. A 527, 4959–4966 (2010).
Wu, X., Tao, N., Hong, Y., Lu, J. & Lu, K. Localized solid-state amorphization at grain boundaries in a nanocrystalline Al solid solution subjected to surface mechanical attrition. J. Phys. D: Appl. Phys. 38, 4140–4143 (2005).
Koike, J., Parkin, D. M. & Nastasi, M. Crystal-to-amorphous transformation of NiTi induced by cold rolling. J. Mater. Res. 5, 1414–1418 (1990).
Wu, Y. Q. & Xu, Y. B. Lattice-distortion-induced amorphization in indented [110] silicon. J. Mater. Res. 14, 682–687 (1999).
Szlufarska, I., Kalia, R. K., Nakano, A. & Vashishta, P. Nanoindentation-induced amorphization in silicon carbide. Appl. Phys. Lett. 85, 378–380 (2004).
Li, M. Defect-induced topological order-to-disorder transitions in two-dimensional binary substitutional solid solutions: a molecular dynamics study. Phys. Rev. B 62, 13979–13995 (2000).
Ovid'ko, I. A. & Reizis, A. B. Effect of elastic distortions on solid-state amorphization at grain boundaries and dislocations. J. Phys. D: Appl. Phys. 32, 2833–2840 (1999).
Kingma, K. J., Meade, C., Hemley, R. J., Mao, H.-K. & Veblen, D. R. Microstrcutral observations of α-quartz amorphization. Science 259, 666–669 (1993).
Yip, S. The strongest size. Nature 391, 532–533 (1998).
Shen, X. X., Lian, J. S., Jiang, Z. H. & Jiang, Q. The optimal grain sized nanocrystalline Ni with high strength and good ductility fabricated by a direct current electrodeposition. Adv. Eng. Mater. 10, 539–546 (2008).
SchiØtz, J., Di Tolla, F. D. & Jacobsen, K. W. Softening of nanocrystalline metals at very small grain sizes. Nature 391, 561–563 (1998).
Chen, H.-P. et al. Embrittlement of metal by solute segregation-induced amorphization. Phys. Rev. Lett. 104, 155502 (2010).
Chen, M. W., McCauley, J. W. & Hemker, K. J. Shock-induced localized amorphization in boron carbide. Science 299, 1563–1566 (2003).
Acknowledgements
We are grateful for financial support of the National key Basic Research and Development Program (grant 2010CB631001) and the National Natural Science Foundation of China (grant 50871046). We deeply appreciate Xiaozhou Liao for his assistance in TEM analysis and fruitful discussions. We thank Xixun Shen for assistance in sample preparation and Shicheng Sun for helping SEM observations.
Author information
Authors and Affiliations
Contributions
J. S. L. designed the project and guided the research. S. H. carried out the experiments and wrote the paper. L. Z. and S. H. conducted TEM and HRTEM observations. J. S. L. and Q. J. revised the paper. All authors contributed to the discussions.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Han, S., Zhao, L., Jiang, Q. et al. Deformation-induced localized solid-state amorphization in nanocrystalline nickel. Sci Rep 2, 493 (2012). https://doi.org/10.1038/srep00493
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep00493
This article is cited by
-
Pt-induced atomic-level tailoring towards paracrystalline high-entropy alloy
Nature Communications (2023)
-
Thermomechanical Processing of Metal Feedstock for Semisolid Forming: A Review
Metallurgical and Materials Transactions B (2018)
-
Investigation of Interfacial Layer for Ultrasonic Spot Welded Aluminum to Copper Joints
Scientific Reports (2017)
-
Synthesis and characterization of ultralong SiC nanowires with unique optical properties, excellent thermal stability and flexible nanomechanical properties
Scientific Reports (2017)
-
Correlation of Fracture Mode Transition of Ceramic Particle with Critical Velocity for Successful Deposition in Vacuum Kinetic Spraying Process
Journal of Thermal Spray Technology (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
