
Abstract
Multiple sclerosis (MS) is an inflammatory, autoimmune disease of the central nervous system. The cause of MS is still unknown but epidemiological and immunological studies have implicated Epstein-Barr virus (EBV), which infects B cells, as a possible etiological agent involved in disease. Of particular interest is EBV latent membrane protein 2A (LMP2A) because previous studies have demonstrated that LMP2A enhances the expansion and differentiation of B cells upon antigen stimulation, revealing a potential contribution of this protein in autoimmunity. Since B cells are thought to contribute to MS, we examined the role of LMP2A in the animal model experimental autoimmune encephalomyelitis (EAE). In this model, transgenic mice in which B cells express LMP2A show increased severity and incidence of disease. This difference was not due to lymphocyte recruitment into the CNS or differences in T cell activation, rather, we show that LMP2A enhances antigen presentation function.
Similar content being viewed by others


Introduction
MS is a chronic inflammatory disease of the central nervous system (CNS) that begins in early adulthood. More than 1 million people worldwide and at least 350,000 people in the United States are affected with MS1. This disease is characterized by focal lesions of demyelination, which leads to episodic or progressive neurological disability1. The cause and mechanisms of MS have yet to be determined, but it is thought to arise via a combination of a genetic susceptibility, tissue damage and environmental factors, such as a viral infection2 that may lead to a break in tolerance.
Infection with EBV, a B-lymphotropic gamma-herpesvirus, is correlated with MS and as such is a leading candidate etiological factor in MS1,3,4. Although the exact relationship between EBV infection and MS is not clearly identified, MS has been associated with latent EBV infection of B cells. As B cells play a fundamental role in immunity and because studies have already outlined the potential role of the humoral arm of the immune system in both MS5,6,7,8 and EAE5,9,10,11, B cell dysregulation through latent persisting viral infection may contribute to autoimmunity.
Latent membrane protein 2A (LMP2A) is an EBV protein expressed during primary and latent infection and has been extensively studied in transgenic models. These data indicate that LMP2A promotes B cell survival, development, proliferation and differentiation12,13,14. Thus, dysregulation of normal B cell function by LMP2A may constitute a mechanism underlying the role of EBV in autoimmunity.
To investigate the hypothesis that LMP2A may contribute to autoimmunity, we utilized our transgenic LMP2A mice (Tg6) that express LMP2A at levels that do not significantly alter B cell development15. Similar to earlier studies using an animal model of systemic lupus erythematosus that determined that LMP2A induces autoreactive B cell activation16, we demonstrate that by enhancing antigen presentation function, LMP2A enhances disease severity in an animal model of MS.
Results
LMP2A increases clinical symptoms of EAE
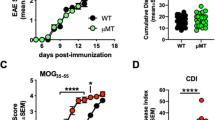
To determine whether the expression of LMP2A in B cells alters the development of EAE, transgenic LMP2A mice and litter mate controls (WT) were immunized with human recombinant myelin oligodendrocyte glycoprotein (rMOG) in CFA and monitored for clinical disease development. Human rMOG was used instead of MOG35–55 peptide because B cells were revealed to be important when immunizing with protein and not peptide11 and with human rMOG and not rat rMOG17. LMP2A mice developed slightly worse disease than did litter mate controls (Figure 1). LMP2A mice have a higher disease incidence and peak score and develop more severe disease, although the day of onset of disease is similar compared to litter mate controls (Table 1).

LMP2A increases clinical symptoms of EAE.
Clinical scores of rMOG/CFA + PTX -induced EAE in LMP2A and litter mate control (WT) mice were collected. A representative of three independent experiments is shown, in which each data point represents the mean and ± SEM of 6 WT and 7 LMP2A mice.
CNS lymphocyte infiltrates do not differ between LMP2A transgenic mice and litter mate controls
To test the hypothesis that differing cell infiltrating profiles between LMP2A mice and litter mate controls could explain the enhanced clinical symptoms of EAE, mononuclear cells were isolated from the CNS and quantified during the pre-clinical (days 7–9), peak (days 15–16) and chronic (days 22–24) stages of EAE. Figure 2a demonstrates the flow cytometric gating strategy used to quantify total cells and lymphocyte subsets. We observed similar numbers of total infiltrating cells in brains (data not shown) and spinal cords of both LMP2A mice and litter mate controls (Figure 2b) as well as in the populations of CD3+CD4+, CD3+CD8+ and CD19+ cells (Figure 2c–e) at all three disease stages.

CNS lymphocyte infiltrates do not differ between LMP2A transgenic mice and litter mate controls.
Mononuclear cells were isolated from spinal cords of LMP2A and litter mate control (WT) mice at pre-clinical, peak and recovery stages of disease. a, Flow cytometric gating strategy used to quantify total cells and lymphocyte subsets. b, Enumeration of infiltrating cells in LMP2A and WT littermates at all three stages of disease using trypan blue exclusion. c-e, Infiltrating cells were stained to identify CD45hiCD19+, CD45hiCD3+CD4+ and CD45hiCD3+CD8+ cells by flow cytometry at pre-clinical (c), peak (d) and chronic (e) stages of EAE. Each number represents a mouse and shown is a representative of 3 (pre-clinical), 4 (peak) and 2 (chronic) experiments. Lines represent the mean of each group.
T Cell Activation in LMP2A transgenic mice and litter mate controls are similar
Due to the lack of differences found in the infiltrating cell populations in the CNS of mice throughout EAE, we next determined if the T cell activation state may explain differences in disease between LMP2A mice and litter mate controls. Initially, bulk cells from lymph nodes and spleens from mice at the pre-clinical, peak and chronic stages of EAE were restimulated with rMOG and proliferation was measured using 3H-thymidine incorporation. Both lymph node cells (data not shown) and splenocytes (Figure 3a) from LMP2A and litter mate control mice proliferated similarly.

T cell activation in LMP2A transgenic mice and litter mate controls (WT) are similar.
a, Splenocytes from primed (solid lines) and naive (dotted lines) mice at peak disease were restimulated with the indicated concentrations of rMOG and proliferation was determined via 3H-thymidine incorporation. b and c, Splenocytes were collected at peak disease and restimulated with the indicated amount of rMOG. Supernatants from 3 days of culture were analyzed for cytokine levels using cytokine bead array (CBA). d and e, Cells from lymph nodes were collected at pre-clinical, peak and chronic EAE and restimulated with MOG35–55 peptide and assessed for the number of IL-2 and IFN-γ secreting cell using ELISPOT detection. The vertical bars represent mean ± SD between triplicate wells. Statistical difference of p = 0.0046 (**) is indicated.
To examine the cytokine profile of cells during EAE, splenocytes were collected at peak disease and restimulated with MOG35–55, an immunodominant determinant of MOG. The cytokine profiles in the culture supernatants were measured, demonstrating no difference in IL-2 (Figure 3b), IFN-γ (Figure 3c), IL-17 (data not shown), or TNF-α levels (data not shown) between LMP2A and litter mates control groups. Additionally, no difference was found in lymph node cells or at other stages of EAE (data not shown).
To further examine the activation status of T cells during EAE, cells were isolated from spleen and lymph nodes at the pre-clinical, peak and chronic stages of EAE and were restimulated with MOG35–55 and assessed for the number of IL-2 and IFN-γ secreting cells. The number of IL-2 secreting cells from spleens (data not shown) and lymph nodes did not differ between LMP2A and litter mate controls groups throughout disease (Figure 3d). However, the number of IFN-γ secreting cells was enhanced at the pre-clinical disease stage in LMP2A mice compared to litter mate controls (Figure 3e).
IgG1 levels are increased in LMP2A transgenic mice when compared to litter mate controls
To determine if increased autoantibody levels could explain the increased disease scores in LMP2A mice, immunoglobulin ELISAs were performed to examine the levels of total rMOG-specific IgG in serum collected from LMP2A mice and litter mate controls at the pre-clinical, peak and chronic stages of EAE. No differences in rMOG-specific IgG between LMP2A mice and litter mate controls were detected at pre-clinical (Figure 4a), peak (data not shown), or chronic (data not shown) EAE. To examine if B cells from mice were secreting differential levels of IgG isotypes, cells from lymph nodes (Figure 4c–e) and spleens (Figure 4f–h) were collected at the pre-clinical, peak and chronic stages of EAE and the number of rMOG-specific IgG1, IgG2a and IgG2b antibody secreting cells (ASCs) were assessed using an ELISPOT assay. The numbers of rMOG-specific IgG2a and IgG2b secreting cells were similar in LMP2A mice and litter mate controls at all stages of EAE. However, in both lymph nodes and spleens, LMP2A mice had increased numbers of rMOG-specific IgG1 secreting cells at the pre-clinical disease stage (Figure 4c, f). The number of ASCs between splenocytes and lymph node cells were similar. As support to the finding of increased numbers of IgG1-secreting cells at pre-clinical EAE, levels of rMOG-specific IgG1 were increased in serum of mice at this stage of disease (Figure 4b).

IgG1 levels are increased in LMP2A transgenic mice when compared to litter mate controls.
rMOG-specific antibody production by B cells of LMP2A mice and litter mates (WT) after rMOG-induced EAE were measured. a-b, ELISA of rMOG-specific IgG (a) and IgG1 (b) in 5-fold dilutions (160X–500,000X) of serum collected from naïve (dotted lines) and pre-clinical EAE (solid lines) mice. Individual mouse serum samples were tested in duplicate and the data is represented as mean ± SD of 2 experiments, each experiment consisting of 2–4 mice per genotype. Statistical differences of p = 0.0156 (*) or p = 0.0051 (**) at the specific dilutions are indicated. c–h, ELISPOT detection of rMOG-specific IgG isotypes produced by lymph node (c–e) and spleen cells (f–h) at pre-clinical, peak and chronic EAE after restimulation with MOG35–55 peptide. Data is graphed as percent wildtype and shown are the means ± SD of 3 (pre-clinical), 4 (peak) and 2 (chronic) experiments. The range of spots detected per 500,000 cells plated were 34–90, 8–43, 15–68 (IgG1), 18–48, 14–23, 47–52 (IgG2a) and 18–46, 18–34, 42–85 (IgG2b) for pre-clinical, peak and chronic stages of EAE, respectively.
Antigen presentation function is enhanced in LMP2A transgenic mice when compared to litter mate controls
Because LMP2A is known to enhance B cell activation13,16, we wanted to determine whether expression of LMP2A increased expression of MHC class II and co-stimulatory molecules in B cells. LMP2A mice were crossed with BCRMOG transgenic mice18. B cells from double transgenic (LMP2A-BCRMOG) and litter mate control mice (WT-BCRMOG) were isolated and cultured with T cells isolated from TCRMOG mice19 and rMOG. Flow cytometric analysis revealed that culture with increased rMOG resulted in increased expression of MHC class II, CD80 and CD86; however, increased expression of these proteins were similar on both LMP2A and litter mate control B cells (Figure 5a). Similarly, T cell activation markers CD25, CD44, CD62L and CD69 were also analyzed on the co-cultured T cells and found to be expressed at control levels. (Figure 5b).

Levels of activation markers are similar between LMP2A transgenic mice and litter mate controls.
T cells purified from TCRMOG mice and B cells purified from LMP2A-BCRMOG (LMP2A) and WT-BCRMOG control (WT) mice were co-cultured with the indicated concentrations of rMOG. Expression of MHCII, CD80 and CD86 on B220+ cells (a) and CD25, CD44, CD62L and CD69 on CD3+ cells (b) were evaluated after 24 hour of culture using flow cytometry.
Since B cells are important professional antigen presenting cells (APCs), we next investigated whether the expression of LMP2A affected the antigen presenting function of B cells. Culture of splenocytes with self-quenching DQ-OVA revealed no difference in antigen processing between control and LMP2A B cells (Figure 6a). However, the proliferation of MOG-specific T cells stimulated with rMOG in co-cultures with irradiated B cells from LMP2A-BCRMOG, as compared to B cells from WT-BCRMOG mice, was significantly enhanced (Figure 6b). This result was observed with two different concentrations of rMOG and at two timepoints and reproducible with two separate preparations of rMOG protein.

Antigen presentation function is enhanced in LMP2A transgenic mice when compared to litter mate controls.
a, Splenocytes from LMP2A and litter mate control mice (WT) were cultured with different concentrations of self-quenching DQ-OVA and FITC signal was measured from CD19+ cells cultured at 37°C (solid lines) and 4°C (dotted lines). b-d, 3H-thymidine uptake by T cells purified from TCRMOG mice and B cells purified from LMP2A-BCRMOG (LMP2A) and WT-BCRMOG (WT) mice were measured after culturing with indicated concentrations of rMOG or MOG35–55 peptide. Cells were cultured for three (b) or four (c and d) days and pulsed with 3H-thymidine for the final 24 hours. Before culturing, B cells were irradiated (b and c) or fixed with paraformaldehyde (d). The data are representative of 2 (a), 5 (b), 3 (c) and 3 (d) independent experiments. Vertical bars represent mean ± SD between triplicate wells. Statistical differences of p<0.05 (*) or p<0.01 (**) are indicated.
To bypass processing of antigen, B cells from LMP2A-BCRMOG and WT-BCRMOG mice were cultured with MOG35–55-specific T cells in the presence of MOG35–55 peptide. LMP2A B cells stimulated increased T cell proliferation as compared to B cells of litter mate controls (Figure 6c). To investigate the need for processing of the MOG35–55 peptide, B cells were fixed with paraformaldehyde to prevent antigen processing but allow peptide loading. Cultures with fixed B cells from LMP2A-BCRMOG and WT-BCRMOG mice induced similar levels of T cell proliferation at two timepoints and three concentrations of antigen (Figure 6d). As expected, culture with rMOG protein and PFA-fixed B cells resulted in nominal T cell proliferation (data not shown). Collectively LMP2A B cells exhibit enhanced capacity to drive activation of naïve T cells.
Discussion
In the current study, we investigated if EBV LMP2A influences autoimmunity using EAE, an accepted murine model of MS. LMP2A is expressed in EBV latent infection and is known to alter normal B cell function. We found that clinical EAE scores were increased in LMP2A mice compared to litter mate controls. This enhancement was not due to do enhanced cell infiltration into the CNS or enhanced T cell activation despite the observation in in vitro models that LMP2A enhances cell migration20,21,22. Interestingly, levels of IFN-γ-producing T cells and rMOG-specific IgG1 secreting cells were increased during the pre-clinical stage of disease. Most significantly, we found that LMP2A enhanced the APC function of B cells ex vivo, which may provide a mechanistic explanation for the contribution of EBV in autoimmune diseases such as MS. We suggest that increased autoreactive T cell activation as a result of LMP2A enhancement of B cell APC function promotes an inflammatory environment and facilitates disease progression during disease initiation. In addition, the increase in IFN-γ at pre-clinical EAE may also contribute to exacerbated disease. Following initiation of disease, the role of EBV LMP2A in altering other aspects of B cell function may be obscured by the robust T cell response, explaining the small differences in the pre-clinical stage of EAE and the modest difference in overall disease course.
The mechanism of how LMP2A enhances the antigen presenting function of B cells remains unknown, although many studies have revealed a role of LMP2A in altering signaling pathways leading to cell dysregulation14,23,24. One possibility is that LMP2A enhances toll-like receptor (TLR) sensitivity of B cells, which could lead to enhanced APC function. Jiang et al. determined that TLR9 stimulation enhanced the APC function of B cells25 and Wang et al. observed that LMP2A B cells were hypersensitive to TLR9 activation16. TLRs and in particular TLR9, have already been implicated in EAE disease progression26,27. Whether expression of LMP2A in B cells causes heightened sensitivities to TLR ligands in our model of EAE and if those changes in sensitivity affect APC function remain interesting questions to address and is the focus of ongoing studies.
Also, despite the lack of differences in expression levels of MHC class II, CD80 and CD86 on B cells of LMP2A and litter mate controls, distribution and concentration of these molecules within lipid rafts, which has been shown to be important in the formation of immunological synapses28, may be altered by LMP2A, resulting in enhanced T cell proliferation. The lack of difference between B cells of LMP2A and litter mate controls when PFA-fixed may be due to the loss of membrane fluidity, thereby disallowing LMP2A to enhance concentration or cross-linkage of BCR complexes.
Alternatively, LMP2A may be involved in the processing and presentation of antigen instead of the initial signaling of the BCR complex and synapse formation. In this case, LMP2A would play a role in the latter steps of antigen processing and presentation, as the endocytic pathway and initial cleavage of antigen is intact in LMP2A B cells, as demonstrated by the uptake of non-specific antigen DQ-OVA. The effect of LMP2A on antigen presentation and processing needs to be further explored, such as processing of protein, trimming of peptide and transport through the Golgi apparatus.
Our initial observation of increased MOG-specific IgG1-secreting cells in spleens and lymph nodes of mice at the pre-clinical stage of disease was surprising due to the popular belief that EAE is considered a TH1 mediated disease and knowing that cytokines produced by TH2 cells such as IL-4 promotes class switching to IgG1 isotypes. However, the presence of IgG1 during or after an inflammatory response has been reported: high titers of IgG1 autoantibodies are detected in the double-transgenic BCRMOG x TCRMOG mice that develop spontaneous opticospinal encephalomyelitis despite a lack of increase in TH2 cytokines29; the oligoclonal immunoglobulins found in the cerebral spinal fluid of MS patients are predominantly IgG130. In our model, LMP2A B cells may encounter increased levels of TH2 cytokines pre-clinically, leading to IgG1 isotype switching. During the remainder of the disease course and with TH1 cytokines, however, B cells from LMP2A and litter mate controls could be encountering the same cytokines, leading to the observed equivalent levels of isotype switching to IgG1, IgG2a and IgG2b.
Previous studies of LMP2A-transgenic mice also indicate a role for this protein in the initial stages of autoimmunity. LMP2A has been shown to bypass tolerance checkpoints23 and enhance TLR sensitivity, as previously mentioned16. These characteristics or another factor due to enhanced B cell signaling13 could lead to enhanced antigen processing and presentation. The contribution of latently infected B cells as antigen presenting cells may be significant in the context of MS, as myelin antigen would be limiting during the initiation of disease and B cells have been shown to be important antigen presenting cells when antigen is limiting31,32.
The contribution of EBV in MS remains controversial and whether our results are reflective of the events in human disease remains unknown. While EBV remains persistently infected in only a small fraction of B cells in an individual33, all B cells in our transgenic mice express LMP2A34. Also, other viral proteins expressed during latency may affect the role of LMP2A in EAE and MS. These issues could begin to be addressed by adoptively transferring known ratios of B cells from LMP2A and litter mate controls into B cell deficient (μMT) mice, as well as using double transgenic mice which B cells express both LMP2A and other latency proteins.
Our finding that LMP2A enhances antigen presentation of B cells leads to a puzzling question of why EBV might enhance host cell APC function. Since mature, naïve B cells require encounter with antigen to survive, infection of these cells leads to a perplexing situation in which a balance between host cell survival and immune evasion must be maintained. While LMP2A enhances the survival of the host cell, other viral factors may be involved in regulating this effect. For example, gp42, a glycoprotein involved in EBV entry, binds MHC class II and thus interferes with presentation of peptide to T cells35. Investigating other EBV proteins in addition to LMP2A will delineate the individual and net effects of viral proteins on APC function. Alternatively, the effects of LMP2A on B cell APC function may be indirect. In response to LMP2A signaling, B cells may have evolved an anti-viral strategy by enhancing immune surveillance. Although B cells normally maintain a balance between immunodeficiency and autoimmunity, infection with EBV may tip the balance of B cells towards promoting autoimmunity.
In conclusion, we demonstrate that LMP2A enhances EAE severity and antibody production at the pre-clinical stage of disease. We also observed that this latency protein enhances B cell APC function. Collectively, these findings provide a novel potential mechanism to explain how latent EBV infection may contribute to autoimmune diseases, including MS. Further studies are required to definitively delineate the mechanism(s) by which LMP2A enhances antigen presentation and how this results in exacerbated disease.
Methods
Mice
LMP2A transgenic mice were created in a C57BL/6 background as previously described. MOGTCR (2D2) mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and have been previously described. MOGBCR (IgH) mice were obtained from the Kuchroo laboratory (Harvard Medical School, Boston, MA). All mice were bred and housed under specific pathogen-free (SPF) conditions in the Center for Comparative Medicine, Northwestern University. All experiments were performed in accordance with institutional and NIH guidelines and approved by the Animal Care and Use Committee at Northwestern University.
MOG peptide and protein
Synthetic peptide MOG35–55 (MEVGWYRSPFSRVVHLYRNGK) was purchased from Genemed Synthesis (San Francisco, CA). Escherichia coli DH5α bacteria expressing the extracellular 125 amino acid domain of human MOG proteins were obtained from the Ruddle laboratory (Yale University School of Medicine, New Haven, CT) and the protein was purified as previously described. Briefly, the bacteria were grown in antibiotic-supplemented Luria-Bertani broth and induced to overexpress hexahistidine-tagged MOG. The protein was then denatured, purified via Ni-NTA Agarose (Qiagen), then refolded.
Induction and Evaluation of EAE
Six- to eight-wk-old C57Bl/6 mice were immunized subcutaneously with 100 μl of an emulsion containing 100 μg of rMOG and 200 μg of Mycobacterium tuberculosis H37Ra (BD Biosciences, San Jose, CA) distributed over three sites on the flank. Mice were injected intraperitoneally with 200 ng of pertussis toxin (PTX) (List Biological Laboratories, Cambell, CA) on days 0 and 2 post-immunization. Clinical scores were given on a scale of 0 to 5 based on the following symptoms: 0 = healthy; 1 = limp tail; 2 = mild hind limb weakness; 3 = severe hind limb weakness; 4 = hind limb paralysis; 5 = moribund. Immunologic analyses were carried out at pre-clinical (days 7–9), peak (days 15–16) and chronic (days 22–24) stages of EAE.
Cell Isolation
Isolation of CNS-infiltrating mononuclear cells from the CNS was performed as similarly described36, by isolating brains and spinal cords from PBS-perfused mice and treated with a solution of 0.2 Wn/ml Liberase R1 (Roche Applied Science), 50 μg/ml DNase I and 25 mM Hepes in balanced salt solution and mashed through wire screens to obtain single cell suspensions. Cells were washed in 5 mM EDTA-Hank’s balanced salt solution before a Percoll (Amersham, Piscataway, NY) gradient was used to isolate mononuclear cells. For spleen and lymph node, tissues were dissociated through wire screens to obtain single cell suspensions. Red blood cells were lysed in 155 mM ammonium chloride. Cells were then filtered through 40 μm nylon mesh. For APC experiments, CD19+ B cells (from LMP2A-BCRMOG and WT-BCRMOG mice) and T cells (from TCRMOG mice) were purified to approximately 90 to 95% purity via magnetic cell sorting (MACS) columns utilizing magnetic beads coated with antibodies specific for CD19 or CD4 (Miltenyi Biotec, Auburn, CA, USA). CD19+ B cells were irradiated or fixed in 1% paraformaldehyde for 8 minutes at room temperature before 100,000 each of CD19+ B cells and T cells were cultured.
Peripheral In Vitro Antigen-Specific Proliferation Assay
Spleens and lymph nodes (cervical, axillary, brachial and inguinal) were harvested and mashed through wire screens. 5 × 105 erythrocyte-free cells per well were incubated in complete RPMI (consisting of RPMI 1640 [cellgro], 10% fetal bovine serum, 50 uM β-mercaptoethanol, 50 U/ml penicillin and 50 μg/ml streptomycin) with MOG35–55 at 37°C in 5% CO2 for 72 hours in 96-well round-bottom microtiter plates, then pulsed with 1 μCi/well 3H-thymidine for the final 24 hours of culture. Uptake of 3H-thymidine was detected using a Topcount Microplate Scintillation Counter. For antigen presentation experiments, cells were also cultured with indicated amounts of rMOG protein and B cells were irradiated with 818 rad or fixed with 1% paraformaldehyde.
Flow cytometric analysis
Single cell suspensions of spleen, lymph node, brain and spinal cord tissue were stained with the following monoclonal rat anti-mouse antibodies: CD3e (PE-Cy7), CD45 (APC), MHCII I-A/I-E (PE), CD80 (FITC), CD86 (APC), CD25 (APC-Cy7), CD62L (APC), CD69 (FITC) from eBioscience (San Diego, CA), or CD4 (FITC), CD8a (APC-Cy7), CD19 (PE), B220 (PerCP), CD3 (PerCP), CD44 (PE) from BD Biosciences (San Jose, CA). 0.5 μl antibody was added per 500,000 cells and incubated at 4°C for 30 minutes in 2% FBS-PBS. For APC experiments analyzing co-stimulatory molecules, cells were first incubated with LIVE/DEAD Fixable Violet Dead Cell Stain from Invitrogen (Carlsbad, CA) to exclude dead cells. Cells were analyzed using a FACSCanto II machine (BD Biosciences) and FlowJo software (Treestar, Ashland, OR).
Enzyme-linked immunosorbent assay (ELISA)
For serum ELISA, serial dilutions of serum were applied to flat-bottom 96-well plates (Thermo Scientific) coated with 0.25 μg/well rMOG and incubated for 1 hour at 37°C after blocking with a solution of Phosphate Buffered Saline, 0.05% Tween-20 and 5% Bovine Serum Albumin (PBST-BSA). 10 ng/well of polyclonal anti-mouse IgG antibody (eBiosciences) or monoclonal anti-mouse IgG1 antibody (clone A85-1) (BD Biosciences) was incubated for 1 hour at 37°C, 1:10,000 dilution of horseradish peroxidase-conjugated streptavidin (Amersham Biosciences) for 30 min. at 37°C and finally TMB One Component HRP Microwell Substrate and 450 nM Liquid Stop Solution (BioFX) were each applied to the wells. Plates were washed with PBST between steps except before application of Stop Solution. A SpectraMax M2 Microplate Readers (Molecular Devices Corporation, Sunnyvale, CA) was used to read the plates at a wavelength of 450 nm. For cytokine ELISA, supernatants from culture were collected and analyzed via standard protocols for Cytometric Bead Array (CBA) (BD Biosciences).
Enzyme-linked immuno-spot assay (ELISPOT)
350 μl, 0.45 µm PVDF UniFilter plates (Whatman, GE Healthcare) were coated with 0.4 μg/well purified antibody against IL-2 (clone JES6-1A12, eBiosciences) or IFN-γ (clone R4-6A2), or 0.25 μg/well rMOG and incubated overnight at 4°C. 500,000 erythrocyte-free cells per well were incubated in complete RPMI for 24 hrs. at 37°C with 5% CO2. For IL-2 and IFN-γ plates, 100 uM MOG35-55 were also added with the cells. After washing, biotinylated antibodies against the following proteins were applied: IL-2 (clone JES6-5H4, eBiosciences), IFN-γ (clone XMG1.2, eBiosciences), IgG1 (clone A85-1), IgG2a[b] (clone 5.7), IgG2b (clone R12-3) and IgM (R6-60.2). The plates were then incubated with an alkaline phosphatase-conjugated anti-biotin antibody (Vector Labs, Burlingame, CA) for 1 hr. at 37°C and developed using AP Conjugate Substrate Kit (Bio-Rad Laboratories, Hercules, CA). Spots were counted on an ELISPOT plate reader (Cellular Technologies Ltd., Shaker Heights, OH). All antibodies are from BD Biosciences unless indicated.
Statistical Analysis
Graphs are presented as mean ± standard error of the mean (SEM) or standard deviation (SD). Statistical analysis was performed with the GraphPad Prism 5.0. Differences between multiple independent experiments were determined with paired, 2-tailed Student’s t-test. Unpaired, 2-tailed Student’s t-test was used in Figure 3e and 4b. Two-way Anova was used in Figure 6b and 6c. P values less than 0.05 were considered significant (*); p values less than 0.01 were considered very significant (**).
References
Lunemann, J. D., Kamradt, T., Martin, R. & Munz, C. Epstein-barr virus: environmental trigger of multiple sclerosis? J Virol 81, 6777–6784 (2007).
Nikbin, B., Bonab, M. M., Khosravi, F. & Talebian, F. Role of B cells in pathogenesis of multiple sclerosis. Int Rev Neurobiol 79, 13–42 (2007).
Giovannoni, G. & Ebers, G. Multiple sclerosis: the environment and causation. Curr Opin Neurol 20, 261–268 (2007).
Thacker, E. L., Mirzaei, F. & Ascherio, A. Infectious mononucleosis and risk for multiple sclerosis: a meta-analysis. Ann Neurol 59, 499–503 (2006).
Cross, A. H., Trotter, J. L. & Lyons, J. B cells and antibodies in CNS demyelinating disease. J Neuroimmunol 112, 1–14 (2001).
Cross, A. H. MS: the return of the B cell. Neurology 54, 1214–1215 (2000).
Hemmer, B., Cepok, S., Nessler, S. & Sommer, N. Pathogenesis of multiple sclerosis: an update on immunology. Curr Opin Neurol 15, 227–231 (2002).
Smith-Jensen, T. et al. Comparison of immunoglobulin G heavy-chain sequences in MS and SSPE brains reveals an antigen-driven response. Neurology 54, 1227–1232 (2000).
Wolf, S. D., Dittel, B. N., Hardardottir, F. & Janeway, C. A. Experimental autoimmune encephalomyelitis induction in genetically B cell-deficient mice. J Exp Med 184, 2271–2278 (1996).
Lyons, J.-A., Ramsbottom, M. J. & Cross, A. H. Critical role of antigen-specific antibody in experimental autoimmune encephalomyelitis induced by recombinant myelin oligodendrocyte glycoprotein. Eur J Immunol 32, 1905–1913 (2002).
Lyons, J. A., San, M., Happ, M. P. & Cross, A. H. B cells are critical to induction of experimental allergic encephalomyelitis by protein but not by a short encephalitogenic peptide. Eur J Immunol 29, 3432–3439 (1999).
Merchant, M., Caldwell, R. G. & Longnecker, R. The LMP2A ITAM is essential for providing B cells with development and survival signals in vivo. J Virol 74, 9115–9124 (2000).
Swanson-Mungerson, M., Bultema, R. & Longnecker, R. Epstein-Barr virus LMP2A enhances B-cell responses in vivo and in vitro. J Virol 80, 6764–6770 (2006).
Portis, T. & Longnecker, R. Epstein-Barr virus (EBV) LMP2A mediates B-lymphocyte survival through constitutive activation of the Ras/PI3K/Akt pathway. Oncogene 23, 8619–8628 (2004).
Caldwell, R. G., Brown, R. C. & Longnecker, R. Epstein-Barr virus LMP2A-induced B-cell survival in two unique classes of EmuLMP2A transgenic mice. J Virol 74, 1101–1113 (2000).
Wang, H. et al. EBV latent membrane protein 2A induces autoreactive B cell activation and TLR hypersensitivity. J Immunol 177, 2793–2802 (2006).
Oliver, A. R., Lyon, G. M. & Ruddle, N. H. Rat and human myelin oligodendrocyte glycoproteins induce experimental autoimmune encephalomyelitis by different mechanisms in C57BL/6 mice. J Immunol 171, 462–468 (2003).
Litzenburger, T. et al. B lymphocytes producing demyelinating autoantibodies: development and function in gene-targeted transgenic mice. J Exp Med 188, 169–180 (1998).
Bettelli, E. et al. Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J Exp Med 197, 1073–1081 (2003).
Pegtel, D. M. et al. Epstein-Barr-virus-encoded LMP2A induces primary epithelial cell migration and invasion: possible role in nasopharyngeal carcinoma metastasis. J Virol 79, 15430–15442 (2005).
Huang, S., Stupack, D., Liu, A., Cheresh, D. & Nemerow, G. R. Cell growth and matrix invasion of EBV-immortalized human B lymphocytes is regulated by expression of alpha(v) integrins. Oncogene 19, 1915–1923 (2000).
Lu, J. et al. Syk tyrosine kinase mediates Epstein-Barr virus latent membrane protein 2A-induced cell migration in epithelial cells. The Journal of biological chemistry 281, 8806–8814 (2006).
Swanson-Mungerson, M. A., Caldwell, R. G., Bultema, R. & Longnecker, R. Epstein-Barr virus LMP2A alters in vivo and in vitro models of B-cell anergy, but not deletion, in response to autoantigen. J Virol 79, 7355–7362 (2005).
Merchant, M. & Longnecker, R. LMP2A survival and developmental signals are transmitted through Btk-dependent and Btk-independent pathways. Virology 291, 46–54 (2001).
Jiang, W. et al. TLR9 stimulation drives naive B cells to proliferate and to attain enhanced antigen presenting function. Eur J Immunol 37, 2205–2213 (2007).
Miranda-Hernandez, S. et al. Role for MyD88, TLR2 and TLR9 but not TLR1, TLR4 or TLR6 in experimental autoimmune encephalomyelitis. J Immunol 187, 791–804 (2011).
Prinz, M. et al. Innate immunity mediated by TLR9 modulates pathogenicity in an animal model of multiple sclerosis. J Clin Invest 116, 456–464 (2006).
Hiltbold, E. M., Poloso, N. J. & Roche, P. A. MHC class II-peptide complexes and APC lipid rafts accumulate at the immunological synapse. J Immunol 170, 1329–1338 (2003).
Krishnamoorthy, G., Lassmann, H., Wekerle, H. & Holz, A. Spontaneous opticospinal encephalomyelitis in a double-transgenic mouse model of autoimmune T cell/B cell cooperation. J Clin Invest 116, 2385–2392 (2006).
Hestvik, A. L. The double-edged sword of autoimmunity: lessons from multiple sclerosis. Toxins 2, 856–877 (2010).
Rock, K. L., Benacerraf, B. & Abbas, A. K. Antigen presentation by hapten-specific B lymphocytes. I. Role of surface immunoglobulin receptors. J Exp Med 160, 1102–1113 (1984).
Lanzavecchia, A. Antigen-specific interaction between T and B cells. Nature 314, 537–539 (1985).
Thorley-Lawson, D. A. Epstein-Barr virus: exploiting the immune system. Nature reviews. Immunology 1, 75–82 (2001).
Caldwell, R. G., Wilson, J. B., Anderson, S. J. & Longnecker, R. Epstein-Barr virus LMP2A drives B cell development and survival in the absence of normal B cell receptor signals. Immunity 9, 405–411 (1998).
Ressing, M. E. et al. Epstein-Barr virus gp42 is posttranslationally modified to produce soluble gp42 that mediates HLA class II immune evasion. J Virol 79, 841–852 (2005).
Miller, S. D., McMahon, E. J., Schreiner, B. & Bailey, S. L. Antigen presentation in the CNS by myeloid dendritic cells drives progression of relapsing experimental autoimmune encephalomyelitis. Ann N Y Acad Sci 1103, 179–191 (2007).
Acknowledgements
RL is the Dan and Bertha Spear Research Professor and is supported by the Public Health Service grants CA133063 and CA73507 from the National Cancer Institute. SDM is the Judy Gugenheim Research Professor and is supported by grants NS-026543 and NS062365 from the National Institutes of Health. We would like to thank members of the Longnecker and Miller laboratory for help in the completion of these studies, in addition to Michelle Swanson-Mungerson and Melissa Brown for review of this manuscript. MOGBCR (IgH) mice were kindly provided by the Kuchroo laboratory (Harvard Medical School). E. coli expressing human rMOG protein were kindly provided by the Ruddle laboratory (Yale University School of Medicine).
Author information
Authors and Affiliations
Contributions
RAC performed all experimental work, wrote the main manuscript text and prepared figures. RL and SDM supervised the project and reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Chang, R., Miller, S. & Longnecker, R. Epstein-Barr virus latent membrane protein 2A exacerbates experimental autoimmune encephalomyelitis and enhances antigen presentation function. Sci Rep 2, 353 (2012). https://doi.org/10.1038/srep00353
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep00353
This article is cited by
-
Developmental endothelial locus-1 is a homeostatic factor in the central nervous system limiting neuroinflammation and demyelination
Molecular Psychiatry (2015)
-
DNA-Directed Assembly of Nanogold Dimers: A Unique Dynamic Light Scattering Sensing Probe for Transcription Factor Detection
Scientific Reports (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
