
Abstract
Febuxostat, a drug recently approved in the US, European Union and Japan for treatment of gout, inhibits xanthine oxidoreductase (XOR)-mediated generation of uric acid during purine catabolism. It inhibits bovine milk XOR with a Ki in the picomolar-order, but we found that it is a much weaker inhibitor of Rhodobacter capsulatus XOR, even though the substrate-binding pockets of mammalian and bacterial XOR are well-conserved as regards to catalytically important residues and three-dimensional structure and both permit the inhibitor to be accommodated in the active site, as indicated by computational docking studies. To clarify the reason for the difference of inhibitory potency towards the two XORs, we performed molecular dynamics simulations. The results indicate that differences in mobility of hydrophobic residues that do not directly interact with the substrate account for the difference in inhibitory potency.
Similar content being viewed by others

Introduction
Xanthine oxidoreductase (XOR) (Fig. 1) plays an important role in the catabolism of purine substrates and is found in a wide range of organisms from bacteria to man1,2,3. All XORs have similar molecular mass and cofactor composition, although the subunit composition is different in eukaryotic and prokaryotic enzymes (Fig. 1). Mammalian XOR is a homodimer with a molecular mass of 290 kDa: each subunit contains one molybdenum cofactor (Moco, molybdopterin), two [2Fe–2S] centers and one FAD center. On the other hand, the bacterial enzyme from Rhodobacter capsulatus (RcXOR) is a (αβ)2 heterotetramer with a molecular mass of 275 kDa. Despite this enzyme’s heterotetrameric nature, the crystal structure of RcXOR proved to be similar enough to its mammalian counterpart so that it was solved by molecular replacement techniques4. The smaller α-subunit comprises the iron/sulfur domain (A1-A153), containing two [2Fe–2S] clusters, the FAD binding domain (A185-462) and a 31-amino-acid extended peptide that links these two domains. The β-subunit is the Moco-binding domain (B1-777). Although the polypeptide composition is different in mammalian and bacterial enzymes, the spatial arrangement of the four redox centers is very similar in all XORs (Fig. 1). The oxidative hydroxylation of purines takes place at the molybdenum center and reducing equivalents thus introduced are transferred via the two [2Fe-2S] centers to the FAD center, where reduction of the physiological electron acceptor, NAD+ or O2, occurs5,6,7,8.

Crystal structures and cofactor distances of mammalian and bacterial XORs.
(a) Crystal structure of bovine milk XOR (bXOR) as a homodimer. (b) Crystal structure of Rhodobacter capsulatus XOR (RcXOR) as a heterotetramer. Subunits are shown in different colors. (c)–(d) Cofactors of each XOR selected interatomic distances are shown for bXOR, (c) and RcXOR (d). All the structures were illustrated by PyMol from the protein data bank codes 1FO4 and 1JRO.
Human XOR is a target of drugs used to treat gout and hyperuricemia. Allopurinol, a hypoxanthine isomer, which was introduced by Elion et al.9, has been widely prescribed as a treatment of gout for more than 40 years. When XOR catalyzes the hydroxylation of allopurinol (Fig. 2a) to oxipurinol (alloxanthine) (Fig. 2b) at the molybdenum center, Mo(VI) is converted to Mo(IV). The reduced enzyme in the Mo(IV) state forms a tightly bound, inactive complex with oxipurinol. The crystal structures of the oxipurinol-bound form of RcXOR and bovine milk XOR (bXOR) have been determined at 3.0 and 2.1Å resolution, respectively and their features are very similar to each other4,10. In each XOR, clear electron density was observed between the N2 atom of oxipurinol and the molybdenum atom. Moreover, hydrogen bonds were observed with Glu802, Arg880 and Glu1261 of bXOR (corresponding to Glu232, Arg310 and Glu730 of RcXOR, respectively). In addition, two phenylalanine residues (Phe914 and Phe1009 of bXOR, corresponding to Phe344 and Phe459 of RcXOR, respectively), that constrain the inhibitor so that the N2 atom of the pyrazole ring projects towards the Mo atom, are also conserved. Those amino acid residues are essential for substrate binding and activation11,12. Thus, it was proposed that the oxipurinol nitrogen atom (N2) replaces the catalytically labile Mo-OH ligand, resulting in mechanism-based inhibition.

Difference of inhibitory effects of febuxostat on bovine and Rhodobacter capsulatus XORs.
(a)–(c): Structures of (a) allopurinol, (b) oxipurinol and (c) febuxostat. (d) Activity in the presence of various concentrations of febuxostat as a percentage of that in the absence of inhibitor.  , bXOR;
, bXOR;  , RcXOR;
, RcXOR;  , RcXOR H198N mutant. (e) Kinetics of xanthine-NAD+ inhibition. Lineweaver-Burk plots of xanthine-NAD+ activity of RcXOR in the presence of febuxostat. Final concentration of XOR (AFR = 401) was 2.4 nM. Final concentrations of febuxotat: ×, no inhibitor;
, RcXOR H198N mutant. (e) Kinetics of xanthine-NAD+ inhibition. Lineweaver-Burk plots of xanthine-NAD+ activity of RcXOR in the presence of febuxostat. Final concentration of XOR (AFR = 401) was 2.4 nM. Final concentrations of febuxotat: ×, no inhibitor;  , 5 µM;
, 5 µM;  , 10 µM;
, 10 µM;  , 15 µM;
, 15 µM;  , 20 µM. (inset) Secondary plots. The Ki value was obtained from secondary plots of slope of the Lineweaver-Burk plot versus inhibitor concentration.
, 20 µM. (inset) Secondary plots. The Ki value was obtained from secondary plots of slope of the Lineweaver-Burk plot versus inhibitor concentration.
On the other hand, febuxostat (Fig. 2c), which was developed as a non-purine selective inhibitor of XOR, has a more potent and longer-lasting urate-lowering effect than allopurinol in mammalian species13,14. Clinical efficacy and tolerance to febuxostat have been confirmed15,16 and the drug is available as Adenuric (EU), Uloric (US), or Feburic (Japan) for the chronic management of hyperuricemia in patients with gout. Febuxostat (Fig. 2c) is a larger molecule than allopurinol (Fig. 2a) and the binding mechanism to XOR is quite different. Febuxostat fills most of the cavity (binding pocket) of XOR17, acting as a structure-based inhibitor via multiple interactions, including ionic bonding of its carboxyl group with Arg880, hydrogen bonding of the nitrogen atom of the thiazole with Glu802 and sandwiching of the thiazole ring between Phe914 and Phe1009 in bXOR.
Structure-based drug design (SBDD) is a rapidly progressing technique for computational drug design, utilizing the three-dimensional (3D) structures of biomolecules obtained by means of X-ray crystallography or NMR spectroscopy. For example, HIV protease inhibitors18 (Nelfinavir, Viracept), a neuraminase inhibitor19 (Zanamivir, Relenza) and Abl tyrosine kinase20 (an anti-cancer drug; STI-571, Gleevec) have been developed with the help of SBDD. Although febuxostat was not developed with the aid of SBDD, the interaction between the inhibitor and the 3D structure of the binding pocket of XOR is crucial to an understanding of the inhibition mechanism17 and SBDD is expected to be an effective approach for further development of inhibitor design for XOR, as well as other enzymes.
In this study, we experimentally found that, among human, bovine and bacterial XORs whose 3D structures are known thus far, the bacterial XOR was only very weakly inhibited by febuxostat, whereas the mammalian XORs were strongly inhibited. These findings are in contrast to the case of allopurinol, which is covalently bound to all the XORs mentioned above and is equally effective on all of them. These facts indicate that the binding mechanism of febuxostat in the substrate-binding pocket is different between the bacterial XOR and mammalian XORs, even though the important residues for catalysis are all conserved and there is enough space for febuxostat to enter the binding pocket of the bacterial XOR. In order to clarify the reason for this difference in inhibitory potency, detailed study of the interaction between febuxostat and the binding pocket is necessary. Molecular dynamics (MD) is a powerful tool to address this issue, because the experimentally observed Ki difference can be examined in detail in terms of the calculated dynamic trajectories of both the drug and the enzyme. We therefore conducted MD calculations, focussing on the Moco domain (∼800 residues) of the XORs (see supplementary Fig. S1) and compared the results with the experimental findings. The MD findings could account well for the difference in inhibitory potency of febuxostat towards the two XORs. We believe such MD calculations can offer novel insights for structure-based drug discovery.
Results
Experiment
We measured the inhibitory potency of febuxostat towards mammalian and bacterial XORs using steady-state enzyme kinetics with xanthine as the substrate (see Methods for details). Figure 2d shows the relative enzyme activity versus log concentration of febuxostat. No inhibition of RcXOR was observed up to 100 nM febuxostat, while bXOR was completely inhibited at that concentration (Fig. 2d). The inhibition patterns were non-competitive in double-reciprocal plots, with a constant Ki value and decreasing Vmax value with increasing concentration of the inhibitor (Fig. 2e). As febuxostat has detergent-like character, it may influence protein structure at concentrations higher than micromolar, which would be consistent with non-competitive inhibition. Whatever the reason, the high Ki value (17.5 μM) for RcXOR, compared with the estimated Ki value of around 0.1 nM for bXOR17 indicated that febuxostat did not interact effectively with the active site at the molybdenum center of RcXOR.
To understand these results, we employed a protein-ligand docking algorithm, the LigandFit21 module of Discovery StudioR 2.5 (Accelrys Inc., California), to predict the position of febuxostat in the binding cavity of bXOR and RcXOR, using the protein data bank (PDB) codes 1FO48 or 1JRO4, respectively. In this algorithm, the receptor (apo-protein) is rigid and the ligand (febuxostat) is flexible. In the case of bXOR, the position of febuxostat was almost identical with that in PDB code 1N5X, which is the crystal structure of febuxostat-included bXOR. Further, the position of febuxostat in the cavity of RcXOR was essentially the same as in the case of bXOR (see Supplementary Fig. S2) and the receptor-ligand binding affinity scores obtained for bXOR and RcXOR were also almost the same, in conflict with the experimental results.
The design of structure-based inhibitors for bXOR often includes hydrogen-bonding interaction between a nitrile (-CN) of the inhibitor and Asn768 of bXOR. Thus, we experimentally mutated His198 to Asn198 in RcXOR to examine the importance of this residue, where His198 of RcXOR corresponds to Asn768 of bXOR. As shown in Figure 2d, there was no significant change in the inhibitory potency of febuxostat, indicating that the hydrogen bond is not important or is not formed in the case of RcXOR, even though hydrogen bond formation seemed feasible based on the docking results described above.
Therefore, the difference in binding affinities for febuxostat cannot be understood in terms of the static 3D structures of the bacterial and mammalian XORs. To address this issue further, we carried out molecular dynamics (MD) simulations of the interactions.
Molecular dynamics simulations
Focusing only on the Moco-binding domains, which contain the ligand-binding region, we carried out MD simulations for both bXOR and RcXOR with the ligand (febuxostat). This truncated system of ∼800 residues (Figs. 3 and Supplementary Fig. S1; see also Methods) is called the Moco-domain5,7. The terminal residues were appropriately processed for the MD simulations (see Methods for detail). As shown below, neglecting the other domains in the model systems does not affect the stability of our MD simulations.

Stability of febuxostat in the binding pocket of bXOR or RcXOR evaluated from MD simulations.
(a) The Moco-binding domain with febuxostat. (b) Enlarged image of febuxostat and Moco; the distance between febuxostat (C19) and MPT (Mo) is indicated by a double-headed red arrow. (c) Distances between febuxostat (C19) and Moco (Mo) during MD simulations of bXOR or RcXOR. The distances in the two simulations of bXOR are plotted in red and orange and those in the case of RcXOR, in blue/cyan.
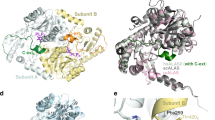
After preparing the Moco domains for bXOR and RcXOR, we inserted febuxostat into these domains using the LigandFit21 module of Discovery StudioR 2.5 (Accelrys Inc., California). The initial position of febuxostat in the binding pocket of bXOR or RcXOR is shown in Supplementary Figure S2a or S2c, respectively. The binding pocket consists of ∼100 residues and the residues around Moco are highly conserved in the two XORs. There are, however, some unconserved residues located far from the Moco site, as shown in Figures 4, 5a and 5b. These residues became the main concern in this study.

Initial and final positions of febuxostat in MD simulations.
The initial position of febuxostat is shown in pink in all the figures (a)–(d). (a)–(b) The final structures of febuxostat after two MD runs of bXOR and its surrounding residues. The residues in black are important for hydrogen-bonding with allopurinol and the residues in red are not conserved between bXOR and RcXOR. (c)–(d) The final structure of febuxostat after two MD runs of RcXOR and its surrounding residues. The residues in black are important for hydrogen-bonding with allopurinol and the residues in blue are not conserved between bXOR and RcXOR.

Comparison of bXOR with RcXOR: interaction between febuxostat and its surrounding three residues.
(a) Hydrophobic residues of bXOR around febuxostat, which was inserted using the docking pose obtained with the protein-ligand docking algorithm. (b) Corresponding RcXOR residues around febuxostat, which was also inserted by using the docking calculation. The residues in black are the same as those in bXOR and those in blue correspond to the ones in red in (a). (c)–(d) Distances between the febuxostat tail atom (C16) and its surrounding three residues in two simulations of bXOR. (e)–(f) Distances between the febuxostat tail atom (C16) and its surrounding three residues in two simulations of RcXOR. Main-chain atoms (N,C,O) are not depicted (except Pro78 of RcXOR) in (a) and (b).
We employed the AMBER software suite22 to simulate the two Moco domains with febuxostat immersed in explicit water molecules (see Methods for detail). For 10 ns MD simulations, it was found that the amino acid backbones fluctuate with 1.4 Å and 1.6 Å RMSD in bXOR and RcXOR, respectively, confirming the stability of the MD simulations (see also the supplementary Fig. S3). Though there is a concern as to whether or not the disordered loop region in RcXOR would affect the results (see Supplementary Fig. S4), we believe that the inclusion of such a loop region in MD simulations would be more likely to increase the fluctuations of the active site, reinforcing our conclusions below.
In two MD runs, febuxostat in bXOR was stable in the binding pocket for 10 ns, while in RcXOR, it was not stable in the binding pocket and after 10 ns the distance between C19 of febuxostat and the Mo atom of Moco was more than 8 Å (Figs. 3, 4 and Supplementary Movie 1–4). The distances reflect the stability of the drug inside the binding pocket, as shown in Figures 3b and 3c. During the MD runs, the distance fluctuated by around 5 Å for bXOR. In the case of RcXOR, however, the distance exceeded 8 Å after 500 ps. The final structures of febuxostat (green) and bXOR are shown in Figure 4a, b, which also depict the initial positions (pink) of febuxostat for comparison. The corresponding structures of RcXOR are shown in Figure 4c, d: febuxostat in RcXOR was found to move out of the binding pocket (to the left-lower side in Fig. 4c and to the left-upper side in Fig. 4d).
Several important residues strongly interacting with febuxostat are also shown in Figure 4a–d: those colored black form hydrogen bonds to febuxostat (also purine derivatives/allopurinol) and are conserved in both bXOR and RcXOR. These residues also play an important role in the hydroxylation of naturally occurring purine derivatives, hypoxanthine and xanthine. On the other hand, the residues colored red in Figure 4a, b or blue in Figure 4c, d are different in the two XORs, indicating that these residues are not associated with the biological hydroxylation function. Because the purine derivatives (hypoxanthine and xanthine) are smaller than febuxostat, the former substrates would not direct contact these residues.
Febuxostat in bXOR is surrounded by hydrophobic residues, except for four hydrogen-bonding residues (Glu802, Arg880, Thr1010 and Asn768 in bXOR). The positions of these residues and febuxostat in bXOR and RcXOR are shown in Figure 5a and 5b, respectively. In the two figures, the residues colored black are conserved in both bXOR and RcXOR and the residues colored red or blue are different in the two XORs; it can be seen that the residues around the head part of febuxostat are conserved, while those around the tail part are not. We believe that this is a key observation for understanding the difference of inhibitory potency and we consider in detail these unconserved residues (Leu648/Pro78, Phe649/Ala79, Val1011/Leu461 and Phe1013/His463 in bXOR/RcXOR) and a hydrogen-bonding residue (Asn768/His198 in bXOR/RcXOR) in the following sections.
Discussion
Allopurinol has been used for remedy of hyperuricemia and gout for more than 40 years and so far febuxostat is the only other drug licensed for these conditions. Since allopurinol is a suicide inhibitor, its potency is much higher than that of competitive inhibitors23. XOR is a highly expressed house-keeping gene product in humans, so potent inhibition of XOR activity is essential to decrease the uric acid level in blood. Febuxostat has a high affinity for XOR, with Ki = 1.2 × 10 −10 M and Ki’ = 1 × 10−9, suggesting that it might be as effective for clinical therapy as allopurinol17,24.
In this article, we focus on the difference in inhibitory potency of febuxostat towards bXOR and RcXOR. Figure 5a, b shows febuxostat and the conserved (black) and unconserved (red or blue) residues in the van der Waals surface representation of these XORs. As shown in Figure 5c, d, the interatomic distances during the molecular dynamics (MD) runs are very stable for bXOR, indicating strong interaction between the tail part of febuxostat and the surrounding residues. On the other hand, the interatomic distances in the case of RcXOR, although its 3D structure is similar to that of bXOR, fluctuate considerably in the two MD runs, as shown in Figure 5e, f. The blue-colored unconserved residues in RcXOR (Fig. 5b) do not appear to contribute to the stabilization of febuxostat inside the binding pocket. Carefully examination of the 3D structure of RcXOR during the MD runs indicated that the packing is slightly looser in the case of RcXOR and a hydrophilic residue (His463) seems to destabilize the binding. On the other hand, the unconserved residues (red) in bXOR are hydrophobic ones and the tail of febuxostat contains several methyl groups, suggesting that hydrophobic interaction between febuxostat and these unconserved hydrophobic residues, Leu648, Phe649 and Phe1013, in bXOR contributes to the strong binding affinity.
Figure 6a, b shows the hydrogen-bonding networks in the two XORs in the wire representation and the distances of these four hydrogen bonds in MD simulations are shown in Figure 6c-f. In the case of bXOR, the four hydrogen bonds remained stable for 10 ns. On the other hand, in the case of RcXOR, the hydrogen bonds involving the bottom residues (His198 and Glu232) were broken faster than the hydrogen bonds involving the other residues (Arg310 and Thr460), implying that instability in the position of the tail part of febuxostat results in breakage of these hydrogen bonds.

Comparison of hydrogen-bonding of bXOR and RcXOR with febuxostat in MD simulations.
(a) Hydrogen-bonding residues around febuxostat of bXOR. (b) Those of RcXOR. (c)–(d) Distances between febuxostat and four hydrogen-bonded residues in two simulations of bXOR. (e)–(f) Distances between febuxostat and four hydrogen-bonded residues in two simulations of RcXOR.
Additional MD simulations were carried out using the same protocol for RcXOR in which His198 is replaced with Asn198 in silico, focusing on the possible hydrogen-bonding interactions between the nitrile (-CN) of febuxostat and some amino acids around febuxostat. It turned out that febuxostat was not stable in the binding pocket, indicating that the mutation does not contribute to the binding affinity for RcXOR, in accordance with the experimental finding using mutant enzyme (Fig. 2d).
The present results demonstrate that MD simulations are a powerful tool to predict the qualitative behavior of a tightly binding inhibitor, in circumstances where conventional docking software may be inadequate. We have shown that even small differences in amino acid sequence, involving residues that do not interact directly with the substrate, can influence the mode of inhibition. Such differences may be extremely important in drug development, for example, allowing us to develop mammalian enzyme-specific inhibitors that would not inhibit the corresponding intestinal bacterial enzyme, or bacterial enzyme-specific inhibitors that would not cause side effects through inhibition of the mammalian enzyme.
Methods
Expression and purification of R. capsulatus XOR in escherichia coli cells
R. capsulatus wild-type XOR was expressed and purified as described previously25. The mutation HB189N was introduced into R. capsulatus XOR by means of PCR mutagenesis. Purification was achieved by nickel-nitrilotriacetic acid chromatography and ion exchange chromatography using Q-Sepharose. To separate Moco-containing XOR from the enzyme lacking the cofactor, affinity chromatography on Sepharose 4B/folate gel was used25. Finally, the protein was purified by size exclusion chromatography. The XOR variants were stored in 50 mM Tris-HCl, pH 7.8, 1 mM EDTA 2.5 mM dithiothreitol.
Purification of bovine XOR
Bovine milk XOR was purified according to Okamoto et al. 17. The concentration of the enzyme was determined spectrophotometrically by using a molar extinction of 37,800 M−1·cm−1 at 450 nm26. The activity-to-flavin ratio (AFR) values of the prepared enzyme were calculated by dividing the absorbance change per minute at 295 nm under standard assay conditions by the enzyme absorbance at 450 nm1.
Enzyme assays
Enzyme assays were carried out at 25°C in 50 mM Tris and 1 mM EDTA, pH 7.8. Routine assay mixtures contained 0.5 mM NAD+ and various concentrations of xanthine or febuxostat. XOR activities were determined by monitoring the absorbance changes at 295 nm as a parameter of uric acid production. Spectrophotometric studies were conducted with a U-3210 spectrophotometer (Hitachi, Tokyo).
Preparation of starting structures
The coordinates for mammalian and bacterial xanthine oxidoreductases (XORs) were taken from Protein Data Bank (PDB) structures 1FO48 and 1JRO4, respectively. The position of febuxostat was determined by a protein-ligand docking algorithm, LigandFit21 module of Discovery StudioR 2.5 (Accelrys Inc., California). To investigate the Moco-binding domain, residues 600–1310 were selected for the bXOR and residues 30–377 & 402–777 for RcXOR. The N- and C-termini of each selected region were capped with an acetyl (ACE) and an N-methyl (NME) group, respectively (besides residue 777 of the bacterial enzyme, which is the biochemical terminus). Although there are missing residues in the bacterial XOR (see PDB: 1JRO) (see Supplementary Fig. S4), this section is far from the binding region and is considered unlikely to be relevant to the enzymatic reaction (see Supplementary Fig. S5). Thus, the missing region was deleted in the model, mimicking the structure of bXOR and its terminal residues were also appropriately processed. Considering hydrogen-bonding between XOR and febuxostat, a hydrogen atom was added to Glu802 of bXOR (Glu232 of RcXOR). All crystal water molecules outside the cut-off range (>8.0 Å) from the regions of interest were removed, but after this procedure, the systems were fully solvated with explicit solvent for the later calculations.
Parameter generation for Moco
To parameterize the partial charges for molybdopterin (Moco), which contains an unconventional Mo atom for molecular dynamics simulations, ab initio quantum chemistry software Gaussian0927 was utilized and the ESP charges were assigned at the level of B3LYP/LANL3DZ/6-31G(d). The general AMBER force field (GAFF) parameters were automatically assigned for the pyranopterin part using the antechamber module. The non-electrostatic parameters for dioxothiomolybdenum were determined empirically and combined with the pyranopterin part. Details of the force field for Moco are given in Supplementary Fig. S6.
Molecular dynamics simulations
All simulations were performed using Amber 11 software22. The Amber ff03 force field was used for proteins and the general Amber force field with the partial ESP charge parameters was assigned for febuxostat. The systems studied were electrostatically neutralized by counter ions in the explicit water (TIP3P) box. The total numbers of atoms in the system were 98,609 and 93,122 for bXOR and RcXOR, respectively (see Supplementary Fig. S1). The systems were minimized for 300 steepest-descent steps and equilibrated for 1 ns with gradually reducing restraints. Finally 10 ns production runs were performed for trajectory analysis with two different initial velocities for each XOR, i.e. two runs for each species. The temperature and the pressure were kept constant by using the Berendsen rescaling method28 and the long-range electrostatic forces were calculated using the particle-mesh Ewald method29.
References
Bray, R. C. Molybdenum iron-sulfur flavin hydroxylases and related enzymes. In: The enzymes XII. (Boyer P. D., editor), Academic Press, New York, 300–419 (1975).
Vogels, G. D. & van der Drift, C. Degradation of purines and pyrimidines by microorganisms. Bacteriol. Rev. 40, 403–468 (1976).
Hille, R. & Massey, V. Molybdenum-containing hydroxylases: Xanthine oxidase, aldehyde oxidase and sulfite oxidase. In: Molybdenum Enzymes. Vol. 7 (Spiro T. G., editor). Wiley-Interscience, New York, 443–518 (1985).
Truglio, J. J., Theis, K., Leimkühler, S., Rappa, R., Rajagopalan, K. V. & Kisker, C. Crystal structures of the active and alloxanthine-inhibited forms of xanthine dehydrogenase from Rhodobacter capsulatus. Structure 10, 115–125 (2002).
Nishino, T. The conversion of xanthine dehydrogenase to xanthine oxidase and the role of the enzyme in reperfusion injury. J. Biochem. (Tokyo) 116, 1–6 (1994).
Hille, R. & Nishino, T. Flavoprotein structure and mechanism. 4. Xanthine oxidase and xanthine dehydrogenase. FASEB J. 9, 995–1003 (1995).
Hille, R. The mononuclear molybdenum enzymes. Chem. Rev. 96, 2757–2816 (1996).
Enroth, C., Eger, B. T., Okamoto, K., Nishino, T., Nishino, T. & Pai, E. F. Crystal structures of bovine milk xanthine dehydrogenase and xanthine oxidase: Structure-based mechanism of conversion. Proc. Natl. Acad. Sci. U.S.A. 97, 10723–10728 (2000).
Elion, G. B., Callahan, S., Nathan, H., Bieber, S., Rundles, R. W. & Hitchings, G. H. Potentiation by inhibition of drug degradation: 6-substituted purines and xanthine oxidase. Biochem. Pharmacol. 12, 85–93 (1963).
Okamoto, K., Eger, B. T., Nishino, T., Pai, E. F. & Nishino, T. Mechanism of inhibition of xanthine oxidoreductase by allopurinol: crystal structure of reduced bovine milk xanthine oxidoreductase bound with oxipurinol. Nucleosides Nucleotides Nucleic Acids. 27, 888–93 (2008).
Yamaguchi, Y., Matsumura, T., Ichida, K., Okamoto, K. & Nishino, T. Crystal structure of human xanthine oxidoreductase mutant, Glu803Val. J. Biochem. (Tokyo). 141, 513–524 (2007).
Okamoto, K., Kawaguchi, Y., Eger, B. T., Pai, E. F. & Nishino, T. Crystal structure of rat D428A mutant, urate bound form. J. Am. Chem. Soc. 132, 17080–17083 (2010).
Osada, Y. et al. Hypouricemic effect of the novel xanthine oxidase inhibitor, TEI-6720, in rodents. Eur. J. Pharmacol. 241, 183–188 (1993).
Komoriya, K. et al. Hypouricemic effect of allopurinol and the novel xanthine oxidase inhibitor TEI-6720 in chimpanzees. Eur. J. Pharmacol. 250, 455–460 (1993).
Becker, M. A. et al. Febuxostat compared with allopurinol in patients with hyperuricemia and gout. N. Engl. J. Med. 353, 2450–2461 (2005).
Burns, C. M. & Wortmann, R. L. Gout therapeutics: new drugs for an old disease. Lancet 377, 165–177 (2011).
Okamoto K, Eger, B. T., Nishino, T., Kondo, S., Pai, E. F. & Nishino, T. An extremely potent inhibitor of xanthine oxidoreductase. Crystal structure of the enzyme-inhibitor complex and mechanism of inhibition. J. Biol. Chem. 278, 1848–1855 (2003).
Kaldor, S. W. et al. Viracept (Nelfinavir Mesylate, AG1343): A Potent, Orally Bioavailable Inhibitor of HIV-1 Protease. J. Med. Chem. 40,3979–3985 (1997).
Varghese, J. N. Development of neuraminidase inhibitors as anti-influenza virus drugs. Drug Develop. Res. 46, 176–196 (1999).
Schindler, T. W., Bornmann, P., Pellicena, W. T., Miller, B., Clarkson. & J. Kuriyan. Structural mechanism for STI-571 inhibition of Abelson tyrosine kinase. Science 289, 1938–1942 (2000).
Venkatachalam, C. M., Jiang, X., Oldfield, T. & Waldman, M. LigandFit: a novel method for the shape-directed rapid docking of ligands to protein active sites. J. Mol. Graph. Model. 21, 289–307 (2003).
Case, D. A. et al. AMBER 11, University of California, San Francisco (2010).
Elion, G. B., Kovensky, A. & Hitchings, G. H. Metabolic studies of allopurinol, an inhibitor of xanthine oxidase. Biochem. Pharmacol. 15, 863–880 (1966).
Takano, Y. et al. Selectivity of febuxostat, a novel non-purine inhibitor of xanthine oxidase/xanthine dehydrogenase. Life Sci. 76, 1835–47 (2005).
Leimkühler, S., Hodson, R., George, G. N. & Rajagopalan, K. V. Recombinant Rhodobacter capsulatus xanthine dehydrogenase, a useful model system for the characterization of protein variants leading to xanthinuria I in humans. J. Biol. Chem. 278, 20802–20811 (2003).
Massey, V., Brumby, P. E. & Komai, H. Studies on milk xanthine oxidase. Some spectral and kinetic properties. J. Biol. Chem. 244, 1682–1691 (1969).
Frisch, M. J. et al. Gaussian 09, Revision A.1 (Gaussian, Inc., Connecticut) (2009).
Berendsen, H. J. C., Postma, J. P. M., van Gunsteren, W. F., Dinola, A. & Haak, J. R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 (1984).
Darden, T. D. York & Pedersen, L. Particle mesh Ewald: An Nlog(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 (1993).
Acknowledgements
This work was supported by Grants-in-Aid for Scientific Research (KAKENHI) 16205021 (T.N.), 23570198 (H.K.) and 22540421(H.F.). This research was also supported by funds for Research and Development of the Next-Generation Integrated Simulation of Living Matter, a part of the Development and Use of the Next-Generation Supercomputer Project of the Ministry of Education, Culture, Sports, Science and Technology (MEXT). S.L. was supported by a Heisenberg fellowship from the Deutsche Forschungsgemeinschaft. H.F. thanks Prof. Akinori Kidera for his critical comments on the initial stage of this work.
Author information
Authors and Affiliations
Contributions
H.K. and T.N. designed the research, S.L. and K.O. conducted the experiments, H.K., H.F. and T.F. conducted the molecular dynamics and docking simulations and H.K., T.N., H.F., T.F., K.O. and S.L. wrote the paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
SUPPLEMENTARY INFORMATION
Supplementary Information
Supplementary Movie 1
Supplementary Information
Supplementary Movie 2
Supplementary Information
Supplementary Movie 3
Supplementary Information
Supplementary Movie 4
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Kikuchi, H., Fujisaki, H., Furuta, T. et al. Different inhibitory potency of febuxostat towards mammalian and bacterial xanthine oxidoreductases: insight from molecular dynamics. Sci Rep 2, 331 (2012). https://doi.org/10.1038/srep00331
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep00331
This article is cited by
-
The significance of serum uric acid level in humans with acute paraquatpoisoning
Scientific Reports (2015)
-
Mechanistic insights into xanthine oxidoreductase from development studies of candidate drugs to treat hyperuricemia and gout
JBIC Journal of Biological Inorganic Chemistry (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
