
Abstract
Multiple sclerosis is an inflammatory demyelinating disease of the central nervous system and a leading cause of neurological disability. The complex immunopathology and variable disease course of multiple sclerosis have limited effective treatment of all patients. Altering the metabolism of immune cells may be an attractive strategy to modify their function during autoimmunity. We examined the effect of inhibiting fatty acid metabolism in experimental autoimmune encephalomyelitis (EAE), a mouse model of multiple sclerosis. Mice treated with an inhibitor of carnitine palmitoyltransferase 1 (CPT-1), the rate-limiting enzyme in the beta-oxidation of fatty acids, showed a reduction in disease severity as well as less inflammation and demyelination. Inhibition of CPT-1 in encephalitogenic T-cells resulted in increased apoptosis and reduced inflammatory cytokine production. These results suggest that disruption of fatty acid metabolism promotes downregulation of inflammation in the CNS and that this metabolic pathway is a potential therapeutic target for multiple sclerosis.
Similar content being viewed by others

Introduction
The neurological disease multiple sclerosis (MS) results in significant disability in affected patients. The pathological hallmarks of MS include infiltration of T cells and macrophages into the CNS, microglial activation, loss of myelin and disruption of motor, sensory and cognitive function1,2,3,4. Although new therapies targeting the immune response have recently been approved or are in clinical trials, these new therapies have significant side effects such as increased risk of opportunistic infections5. Therefore, therapies are needed that specifically target the inflammatory response in the CNS without global immune impression.
Energy production to meet the demands of cellular function is provided by multiple metabolic pathways which use substrates such as glucose (glycolysis) or fatty acids (beta-oxidation) to produce ATP6. Beta-oxidation of fatty acids occurs in the mitochondria which necessitates the importation of fatty acyl groups from the cytosol through the outer and inner mitochondrial membranes7. However, the mitochondrial membrane is impermeable to activated acyl-CoA molecules. Therefore, these molecules must be shuttled through the outer membrane via their conjugation to the quaternary ammonium compound, carnitine, catalyzed by the enzyme CPT-1. In this reaction the acyl group from CoA is transferred to carnitine enabling the importation of acylcarnitines through the outer mitochondria membrane. Subsequently, acylcarnitines are transported through the mitochondrial matrix and converted back to acyl-CoA molecules by a translocase and CPT-2. The conversion of acyl-CoA to acylcarnitine by CPT-1 is the rate limiting step of beta-oxidation and is regulated by the cellular energy status of the cell through the production of its endogenous inhibitor malonyl-CoA; therefore, this reaction serves as an important regulator of cell metabolism7,8.
Several studies indicate a role for fatty acid oxidation in supporting immune cell function. It was recently shown that T cells undergoing metabolic stress such as during the transition from effector to memory T cells, also switch from primarily using glucose as an energy source to relying on fatty acids9. Furthermore, T cells that induce graft vs. host disease (GvHD) show increased levels of acylcarnitine species and a metabolic switch to mitochondrial fatty acid metabolism10. In humans, peripheral blood mononuclear cells isolated from patients with infection show increased levels of acylcarnitines11, indicating that immune-cell activation could increase the use of fatty acids for metabolic fuel.
We sought to examine the role of beta-oxidation during experimental autoimmune encephalomyelitis by using ethyl 2-[6-(4-chlorophenoxy)hexyl]oxirane-2-carboxylate (etomoxir), an irreversible inhibitor of CPT-1. We describe a role for beta-oxidation in promoting CNS inflammation and regulating encephalitogenic T cell responses.
Results
The normal CNS relies primarily on glucose as a major energy source. Although astrocytes express proteins involved in beta-oxidation, they do not utilize fatty acids as a major energy source in nonpathological conditions12,13. In contrast, under conditions of metabolic stress such as rapid proliferation, immune cells have the ability to utilize alternative substrates such as fatty acids10,14. However, it is currently unknown whether inflammatory cells infiltrating into the central nervous system continue to use glycolysis or are able to utilize alternate metabolic programs such as fatty acid oxidation to generate ATP.
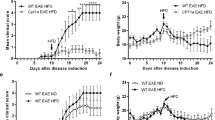
The use of fatty acids as substrates to fuel energy production begins with their importation into the mitochondria via conjugation to carnitine by the enzyme CPT-1 and this is the rate-limiting step of beta-oxidation (Fig. 1). Therefore, we sought to examine the contribution of fatty acid oxidation to the inflammatory response in the CNS by treating mice with the CPT-1 inhibitor, etomoxir. Modulators of fatty acid metabolism such as etomoxir have recently been tested in human phase II clinical trials for congestive heart failure and preclinical evidence also suggests that they may be useful in the treatment of psoriasis15,16. EAE was induced with a peptide from myelin oligodendrocyte glycoprotein (MOG35–55) and mice were administered etomoxir (15 mg/kg i.p.) or vehicle after disease priming. Vehicle-treated mice showed ascending paralysis beginning at approximately day 10, with hind-limb paralysis evident by day 20. However, mice that received two doses of etomoxir displayed no or mild disease symptoms throughout the same time course (Fig. 2a and Supplementary Movies 1–5). CFA-immunized controls displayed no disease and etomoxir treatment of CFA controls did not alter this phenotype (Supplementary Fig. S1 online).

Importation of Fatty Acids into the Mitochondria.
The first step in the beta-oxidation of fatty acids is the transfer of acyl groups from CoA to carnitine due to the inability of acyl-CoA molecules to cross the mitochondrial membrane. Carnitine palmitoyltransferase 1 (CPT 1), located on the outer mitochondrial membrane, transfers the acyl group from CoA to carnitine. This is the rate limiting step in beta-oxidation. Acylcarnitine and free carnitine are subsequently exchanged across the inner mitochondrial membrane by carnitine-acylcarnitine translocase. The acyl group is then transferred back to CoA by carnitine palmitoyltransferase II (CPT-II) on the matrix side of the inner membrane and enters into the beta-oxidation cycle. Etomoxir inhibits CPT I.

CPT-1 Inhibition Ameliorates Disease Activity During EAE.
(a) EAE was induced with MOG peptide. After disease induction, mice received 2 intraperitoneal injections of etomoxir 15 mg/kg on days 8 and 15 (arrows; (○)) while control mice were injected at the same time points with vehicle alone (•) (n = 4 mice per group; data are representative of three independent experiments). Disease was scored as follows; 0 = no disease, 1 = limp tail, 2 = hind- limb paresis, 3 = hind-limb paralysis, 4 = hind/forelimb paralysis and 5 = death. Etomoxir-treated-mice show decreased disease compared to controls (*p<0.05). (b–i) Fluorescence immunohistochemistry of inflammatory lesions in spinal cords from vehicle-treated (B–E) and etomoxir-treated EAE mice (f–i). Sections are stained with FITC-conjugated anti-CD11b to detect macrophages and activated microglia (b, f) and PE-conjugated anti-CD4 to detect CD4+ T-cells (c, g) and counterstained with DAPI (d, h). Merged panels are shown in (e) and (i). Scale bar is 50 microns.
Next we examined the CNS pathology in vehicle-treated and etomoxir-treated mice. To identify lesion-associated CD4+ T cells and CD11b+ macrophages/microglial cells, immunohistochemistry was performed on spinal cords and the extent to which etomoxir treatment decreased the inflammatory response in EAE was determined. EAE mice that did not receive the drug displayed robust infiltration of inflammatory cells into the white matter regions of the spinal cord. In contrast, etomoxir-treated mice displayed a reduced immune cell infiltration in the CNS with few macrophages, activated microglia, or T cells present (Fig. 2b–i and Supplementary Fig. S2 online).
Etomoxir-treated mice displayed less severe inflammation in the spinal cord and this coincided with a reduction in clinical symptoms. Therefore, we examined whether the improved clinical course resulted in less myelin damage in the CNS. Spinal cords from etomoxir or vehicle-treated mice were stained for myelin basic protein and the extent of demyelination was compared (Fig. 3). In vehicle-treated mice, areas of myelin loss coincided with regions of increased cellularity (Fig. 3a–c). In contrast, etomoxir-treated mice had intact myelin sheaths suggesting that a reduction in inflammation prevented myelin destruction (Fig. 3d–f).

Demyelination is Reduced by Etomoxir Treatment during EAE.
Fluorescent immunohistochemistry for myelin basic protein in the spinal cords of vehicle-treated (a–c) or etomoxir-treated (d–f) mice. Prominent areas of myelin pallor are seen in vehicle treated mice (a) that coincide with areas of increased cellularity (b). Merged panel is shown in C. Spinal cords from etomoxir-treated mice show intact myelin (d) along with decreased cellular infiltration (e). Merge of myelin staining and DAPI is shown in F. Magnification is 200x and scale bar is 50 microns.
Etomoxir reduced inflammation and demyelination in the CNS of treated mice; therefore, we investigated whether inhibition of beta-oxidation altered the phenotype of myelin-specific T cells. The effect of etomoxir on T cell cytokine production and apoptosis was examined under conditions that either favored glycolysis, or reduced glucose conditions to induce metabolic stress to favor fatty acid metabolism. Encephalitogenic, MOG-specific T cells were cultured in medium containing high or low concentrations of glucose and cytokine production was measured in the presence or absence of etomoxir. MOG-specific T-cells cultured in IL-12 exhibit a T-helper 1 (Th1) phenotype characterized by secretion of interferon-gamma (IFN-γ). Under high glucose culture conditions, T cells stimulated with MOG35–55 peptide produced high levels of IFN-γ and little IL-4, consistent with a Th1 phenotype exhibited by these cells17. Etomoxir treatment did not alter this profile and no significant difference in IFN-γ production was seen in the presence or absence of drug (Fig. 4a). In contrast, T cells cultured in low glucose medium showed a significant reduction in IFN-γ production in the presence of etomoxir, without concomitant production of the Th2-associated cytokine IL-4 (Fig. 4a). The fact that encephalitogenic T-cells are not skewed toward a Th2 phenotype indicates that the anti-inflammatory effect of etomoxir is on Th1-associated functions of the T-cells, rather than off-target effects modulating T helper cell differentiation. Although MOG-specific T cells were cultured under conditions that skewed the cells to a Th1 phenotype, IL-17 production was also measured after culturing the cells in high and low glucose medium with or without etomoxir (Supplementary Fig. S3 online). Very low levels of IL-17 were detected in high glucose conditions as expected; however, interestingly no IL-17 production was detected when cells were cultured in low glucose. This data is consistent with a previous report that indicates that Th17 cells rely on the glycolytic pathway to promote their differentiation18.

Etomoxir Reduces Pro-Inflammatory Cyokine Production and Increases Apoptosis of MOG Specific T Cells.
(a) Production of IL-4 (black bars) and IFN-γ (grey bars) by MOG-stimulated splenocytes cultured under high or low glucose conditions in the presence of etomoxir or vehicle. Data is represented as mean ± S.D. *p < 0.05. (b) APO-BrdU labeling of cell apoptosis in MOG-stimulated T cells cultured in low (grey bars) or high (black bars) glucose medium. Positive and negative control cells are a lymphoma cell line supplied by the manufacturer. Data is representative of three independent experiments and plotted as mean ± S.D., * p < 0.05. Fluorescence immunohistochemistry for DAPI (c), Apo-BrdU (d) and merge (e) in the spinal cord of a vehicle-treated mouse and DAPI (f), APO-BrdU (g) and merge (h) in the spinal cord of an etomoxir-treated mouse. Increased apoptosis is seen after etomoxir treatment. Scale bar is 50 microns and magnification is 200x. (I) Quantitation of Apo-BrdU fluorescence in spinal cords of vehicle-treated (grey bars) and etomoxir-treated (black bars) mice. Data is expressed as area of Apo-BrdU fluorescence/total spinal cord area (mean ± S.D.).
We then examined whether etomoxir decreased inflammation by promoting apoptosis in activated T cells. Recently, it has been shown that leukemia cells undergo a metabolic switch from glycolysis to fatty acid oxidation and that inhibition of this pathway with etomoxir sensitizes them to induction of apoptosis19. Apoptosis was measured in MOG-specific T cells cultured in high or low glucose medium and subsequently treated with etomoxir or vehicle. Etomoxir had no effect on T cells cultured in high glucose, indicating that antigen-specific T cells retain the ability to perform glycolysis under these conditions. These results support the hypothesis that glycolysis predominates in lymphoid organs where glucose is not limited. In contrast, there was a significant increase (p < 0.05) in apoptosis in etomoxir-treated cultures stimulated with antigen under low glucose conditions (Fig. 4b). Furthermore, APO-BrdU labeling of apoptotic cells in the CNS showed increased evidence of cell death in etomoxir-treated mice (Fig. 4c–i). In addition to promoting apoptosis, etomoxir may also affect the ability of T cells to proliferate. During in vitro stimulation of T cells with MOG antigen T cell numbers increased over the incubation period in high glucose conditions in the presence of etomoxir, but smaller cell numbers were recovered from T cells cultured in low glucose in the presence or absence of etomoxir (data not shown). This indicates that etomoxir may affect multiple T cell functions during metabolic stress and future studies will address whether proliferation or differentiation may be affected. Together these data indicate that etomoxir treatment increases apoptosis of activated, MOG-specific T cells and reduces inflammatory cytokine production under conditions of competition for glucose as an energy substrate; a condition that may occur in the lesion microenvironment within the CNS. The decrease in CNS inflammation seen in vivo may result from the action of etomoxir on MOG-specific T cells.
Discussion
This study examines a link between fatty acid oxidation and autoimmune inflammation in the CNS. Inhibition of CPT-1 resulted in less severe EAE, reduced inflammation and demyelination. Furthermore, inhibition of fatty acid oxidation disrupted encephalitogenic T cell function. Our study points to a pro-survival role for beta-oxidation in encephalitogenic T cells. We directly measured levels of beta-oxidation in encephalitogenic T cells (data not shown) and are pursuing the effect of etomoxir on this pathway.
Several recent studies have demonstrated an increased use of fatty acid substrates as metabolic fuel to promote inflammatory responses in psoriasis, graft vs. host disease and infection10,11,16. This suggests that the high metabolic demands of effector functions such as proliferation and cytokine production necessitates the use of multiple metabolic substrates. Therapies that manipulate upregulated metabolic pathways may prove effective anti-inflammatory therapies, because their blockage would specifically target cell populations responsible for promoting autoimmunity.
Although our studies address the role of etomoxir in encephalitogenic T cells, the anti-inflammatory effect of etomoxir in EAE may be due to multiple factors. Beta-oxidation may be a pro-survival mechanism for antigen-specific or innate inflammatory cells under metabolic stress in the CNS environment. Furthermore, it is unknown how the balance between glycolysis and fatty oxidation is regulated by inflammatory cells in the context of inflammation in the central nervous system. Using 2-[18F]fluoro-2-deoxy-D-glucose and PET imaging, increased glycolysis was detected in the CNS of mice with EAE, indicating increased metabolic demands in inflammatory lesions20. However, the specific cell types responsible for the increased uptake of glucose were not determined. Therefore, it is likely that multiple metabolic pathways are required for energy generation not only by inflammatory cells, but also resident CNS cells within the lesions. Conversely, increased acylcarnitine levels similar to those observed in GVHD or infection10,11 might be toxic in the CNS. Acylcarnitines have been postulated to cross the plasma membrane through an unknown mechanism and previous studies have shown that accumulation of these species during ischemia in the heart leads to disruption of membrane integrity and increases in intracellular calcium21,22. We cannot rule out a detrimental effect of pathway-associated substrates on CNS physiology that may be targeted by etomoxir and this treatment may exert an anti-inflammatory effect on multiple points along this metabolic pathway. Future studies of the roles of different metabolites within the pathway are likely to provide insights into the complex pathogenesis of CNS inflammation.
In this study we identified the beta-oxidation of fatty acids as a metabolic pathway involved in the pathogenesis of EAE. Therapeutic inhibition of one of the primary enzymes involved in acylcarnitine catalysis during fatty acid beta-oxidation, CPT-1, reduced disease symptoms and inflammation in the CNS. These results demonstrate that fatty acid metabolism plays a role in sustaining the inflammatory response within the CNS and provides a new target for therapeutic development.
Methods
Mice
All animals were housed according to institutional animal care and use committee (IACUC) guidelines. Adult female C57BL/6J mice were obtained from the rodent breeding colony maintained by the Scripps Research Institute (La Jolla, CA), or purchased from Charles River Labs and maintained at the University of California, San Diego. EAE was induced as follows; mice received two subcutaneous injections in the flank of 100 μg of peptide derived from myelin oligodendrocyte glycoprotein (MOG 35–55) (AnaSpec, San Jose, CA) on days 0 and 7 along with two intraperitoneal injections of 250 ng pertussis toxin (List Biologicals, Campbell, CA) on days 0 and 2. Mice were scored according to disease severity 0 = -no disease, 1 = limp tail, 2 = hind-limb paresis, 3 = hind-limb paralysis, 4 = hind/forelimb paralysis and 5 = death.
Immunohistochemistry and confocal microscopy
Mice were deeply anesthetized with halothane and intracardially perfused with ice cold PBS. Following perfusion, brain and spinal cord were dissected, frozen in OCT mounting medium (Tissue Tek, Torrance, CA) and 10 µm cryosections were prepared. Inflammatory lesions were identified in tissue sections using FITC conjugated CD11b to detect macrophages and activated microglial cells (eBioscience, San Diego, CA). T-cells were detected using PE-conjugated anti-CD4. Myelin was stained using SMI-99 (Covance, Emeryville, CA) followed by Alexa fluor 488 anti-mouse IgG2b secondary (Invitrogen, Carlsbad, CA). DAPI was used as a counter stain. Images were taken with the Bio-Rad (Zeiss) Radiance 2100 Rainbow laser scanning confocal microscope (Hercules, CA) or an Olympus FluoView1000 confocal microscrope (Center Valley, PA) and Image J analysis software (http://rsb.info.nih.gov/ij/).
Etomoxir treatment
Etomoxir (Sigma, St. Louis, MO) was dissolved in sterile water. Mice received intraperitoneal injections of 15 mg/kg in a volume of 100 μl starting on day 8 after EAE induction and either weekly or every other day for two weeks. Control (vehicle-treated) mice received sterile water only.
T cell cultures
C57BL/6 mice were immunized with 200 μg MOG 35-55 peptide s.c. and 14 days later splenocytes were isolated and cultured at a concentration of 6 × 106 cells/ml in DMEM containing high (4.5 g/L) or low (1 g/L) glucose, 10% FBS 50 μg/ml MOG peptide and 25 ng/ml IL-12 (R&D systems, Minneapolis, MN) in the presence of vehicle or 100 μM etomoxir. 72 hours later cells were harvested for APO-BrdU staining and supernatants collected for IL-4, IL-17 and IFN-γ ELISA.
APO-BrdU staining
Apoptosis was detected in MOG-stimulated T cells using the APO-BrdU kit (Invitrogen, Carlsbad, CA) according to manufacturer’s instructions. Briefly, cells were fixed in 1% paraformaldehyde followed by incubation in 70% ethanol at −20°C for 18 hours. DNA was labeled with BrdU for 60 minutes at 37°C followed by detection with Alexa fluor 488-labeled anti-BrdU (Invitrogen, Carlsbad, CA) and counterstained with PI/Rnase A staining buffer. Samples were analyzed at the UCSD Center for AIDS Research flow cytometry core facility. Tissue sections were fixed in 4% formaldehyde, washed in PBS and permeabilized with proteinase K. Sections were labeled the BrdU for 1.5 hours at 37°C and labeling detected with Alexa Fluor 488 anti-BrdU antibody followed by counterstaining with DAPI. Slides were mounted in aquamount and imaged as previously described.
ELISA
Supernatants were harvested from MOG T cell cultures 72 hours after peptide stimulation and analyzed using IL-4, IFN-γ and IL-17 ELISA kits according to manufacturer’s instructions. (eBioscience, San Diego, CA).
Statistics
Disease scores are expressed as means ± standard error of the mean (SEM). Statistical analysis was performed with two-tailed Student’s t tests for unpaired data. For the animal studies, disease scores were analyzed using the Mann-Whitney U test (GraphPad Prism), where a p value of <0.05 was considered as statistically significant.
References
Korn, T. et al. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med 13, 423–431 (2007).
Baxter, A. G. The origin and application of experimental autoimmune encephalomyelitis. Nat Rev Immunol 7, 904–912 (2007).
Lane, T. E. et al. A central role for CD4(+) T cells and RANTES in virus-induced central nervous system inflammation and demyelination. J Virol 74, 1415–1424 (2000).
Kornek, B. et al. Multiple sclerosis and chronic autoimmune encephalomyelitis: a comparative quantitative study of axonal injury in active, inactive and remyelinated lesions. Am J Pathol 157, 267–276 (2000).
De Jager, P. L. & Hafler, D. A. New therapeutic approaches for multiple sclerosis. Annu Rev Med 58, 417–432 (2007).
Buchakjian, M. R. & Kornbluth, S. The engine driving the ship: metabolic steering of cell proliferation and death. Nat Rev Mol Cell Biol 11, 715–727 (2010).
Houten, S. M. & Wanders, R. J. A general introduction to the biochemistry of mitochondrial fatty acid beta-oxidation. J Inherit Metab Dis 33, 469–477 (2010).
Hostetler, H. A. et al. Acyl-CoA binding proteins interact with the acyl-CoA binding domain of mitochondrial carnitine palmitoyl transferase I. Mol Cell Biochem, (2011).
Pearce, E. L. et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 460, 103–107 (2009).
Gatza, E. et al. Manipulating the bioenergetics of alloreactive T cells causes their selective apoptosis and arrests graft-versus-host disease. Sci Transl Med 3, 67ra68 (2011).
Cress, A. P., Fraker, P. J. & Bieber, L. L. Carnitine and acylcarnitine levels of human peripheral blood lymphocytes and mononuclear phagocytes. Biochim Biophys Acta 992, 135–139 (1989).
Abdul Muneer, P. M., Alikunju, S., Szlachetka, A. M. & Haorah, J. Methamphetamine inhibits the glucose uptake by human neurons and astrocytes: stabilization by acetyl-L-carnitine. PLoS ONE 6, e19258 (2011).
Mosconi, L., Pupi, A. & De Leon, M. J. Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer's disease. Ann N Y Acad Sci 1147, 180–195 (2008).
Pedersen, L. B., Nashold, F. E., Spach, K. M. & Hayes, C. E. 1,25-dihydroxyvitamin D3 reverses experimental autoimmune encephalomyelitis by inhibiting chemokine synthesis and monocyte trafficking. J Neurosci Res 85, 2480–2490 (2007).
Holubarsch, C. J. et al. A double-blind randomized multicentre clinical trial to evaluate the efficacy and safety of two doses of etomoxir in comparison with placebo in patients with moderate congestive heart failure: the ERGO (etomoxir for the recovery of glucose oxidation) study. Clin Sci (Lond) 113, 205–212 (2007).
Caspary, F. et al. A new therapeutic approach to treat psoriasis by inhibition of fatty acid oxidation by Etomoxir. Br J Dermatol 153, 937–944 (2005).
Chang, T. T., Jabs, C., Sobel, R. A., Kuchroo, V. K. & Sharpe, A. H. Studies in B7-deficient mice reveal a critical role for B7 costimulation in both induction and effector phases of experimental autoimmune encephalomyelitis. J Exp Med 190, 733–740 (1999).
Shi, L. Z. et al. HIF1{alpha}-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med 208, 1367–1376 (2011).
Samudio, I. et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J Clin Invest 120, 142–156 (2010).
Radu, C. G., Shu, C. J., Shelly, S. M., Phelps, M. E. & Witte, O. N. Positron emission tomography with computed tomography imaging of neuroinflammation in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A 104, 1937–1942 (2007).
Yamada, K. A., Kanter, E. M. & Newatia, A. Long-chain acylcarnitine induces Ca2+ efflux from the sarcoplasmic reticulum. J Cardiovasc Pharmacol 36, 14–21 (2000).
Ford, D. A., Han, X., Horner, C. C. & Gross, R. W. Accumulation of unsaturated acylcarnitine molecular species during acute myocardial ischemia: metabolic compartmentalization of products of fatty acyl chain elongation in the acylcarnitine pool. Biochemistry 35, 7903–7909 (1996).
Acknowledgements
LPS was supported by postdoctoral fellowships from National Institutes of Health/National Institute of Neurological Disorders and Stroke (1 F32 NS068015-01A1 and T32 NM041219). MM was supported by National Institutes of Health RR025774, P30MH062261 and P30AI036214. Confocal microscopy was supported by the UCSD Neuroscience Microscopy Shared Facility Grant P30 NS047101. We thank J. Young for critical reading of the manuscript.
Author information
Authors and Affiliations
Contributions
LPS conceived of and designed the experiments, performed the experiments, analyzed data and wrote the manuscript. MM conceived of and designed the experiments, analyzed data and wrote the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Movie 1
Supplementary Information
Supplementary Movie 2
Supplementary Information
Supplementary Movie 3
Supplementary Information
Supplementary Movie 4
Supplementary Information
Supplementary Movie 5
Supplementary Information
Shriver Supplemental Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Shriver, L., Manchester, M. Inhibition of fatty acid metabolism ameliorates disease activity in an animal model of multiple sclerosis. Sci Rep 1, 79 (2011). https://doi.org/10.1038/srep00079
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep00079
This article is cited by
-
GC–MS based untargeted metabolomics reveals the metabolic response of earthworm (Eudrilus eugeniae) after chronic combinatorial exposure to three different pesticides
Scientific Reports (2023)
-
Venlafaxine’s effect on resilience to stress is associated with a shift in the balance between glucose and fatty acid utilization
Neuropsychopharmacology (2023)
-
Inhibition of carnitine palmitoyl-transferase 1 is a potential target in a mouse model of Parkinson’s disease
npj Parkinson's Disease (2023)
-
Fatty acids role in multiple sclerosis as “metabokines”
Journal of Neuroinflammation (2022)
-
Thinking outside the box: non-canonical targets in multiple sclerosis
Nature Reviews Drug Discovery (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
