
Key Points
-
Describes the clinical and dental features of Hypophosphatasia.
-
Provides a differential diagnosis for early loss of primary teeth.
-
Encourages GDP's to consider Hypophosphatasia when encountering patients with early loss of primary teeth.
-
Discusses new treatments available which may impact on the prognosis and treatment of patients with Hypophosphatasia
Abstract
Hypophosphatasia (HPP) is an inherited metabolic disorder that results in poorly mineralised bones and teeth. Clinical symptoms vary widely from mild dental anomalies to fatal fetal defects. The most common dental symptoms include exfoliation of the primary incisors before the age of three with little or no root resorption, large pulp chambers, alveolar bone loss and thin dentinal walls. There is generally minimal periodontal inflammation associated with the bony destruction and tooth loss. The general dental practitioner is usually the first clinician to spot signs of the milder forms of HPP. Patients diagnosed with dental symptoms in childhood can go on to develop significant morbidity in middle age with chronic bone pain and stress fractures of the long bones. The primary dental care clinician is the key to early diagnosis of such cases, whether they present in childhood or adulthood. Emerging enzyme replacement therapy has considerably changed the landscape of the disease, resulting in astonishing improvements in bone mineralisation and a significant reduction in mortality and morbidity. It is increasingly likely that primary and secondary care clinicians will treat patients with the severe forms of HPP, who would previously not have survived infancy.
Similar content being viewed by others


Background
Hypophosphatasia (HPP) is a rare inherited metabolic disorder, caused by defects in the gene encoding tissue non-specific alkaline phosphatase enzyme (TNSALP), resulting in impaired mineralisation of bones and teeth.
Clinical symptoms
HPP tends to manifest at different ages, depending on the severity of the phenotype. It is divided into six forms.1
These are:
-
1
Perinatal (previously perinatal lethal)
-
2
Perinatal benign
-
3
Infantile
-
4
Childhood
-
5
Adult
-
6
Odonto-hypophosphatasia.
Perinatal or perinatal lethal HPP generally presents in utero or at birth. There is a near complete lack of skeletal mineralisation with short limbs and/or fractures present. These infants usually require ventilation from birth as they have hypoplastic lungs and a rachitic chest. The poorly mineralised ribs bend easily and fail to allow adequate chest expansion and effective gas exchange leading to respiratory failure. Pyridoxine (vitamin B6) deficient seizures and disturbances of the calcium and phosphate metabolism are also seen.1,2,3
Perinatal benign is a very rare form of HPP. Infants are diagnosed on ultrasound as having bowed legs and variable levels of skeletal mineralisation that undergoes spontaneous improvement in the third trimester of pregnancy. They continue to improve post-natally and seem to manifest only mild symptoms by later life.3,4
Infantile HPP tends to present within the first six months of life with a failure to thrive and increasing respiratory complications and may also present with seizures.
Craniosynostosis is seen in both perinatal and infantile HPP and can result in increased intracranial pressure, which may necessitate cranial surgery.1,5 Hypercalcaemia is nearly always seen, resulting in hypercalciuria and nephrocalcinosis.
Prior to the very recent introduction of enzyme replacement therapy, more than 50% of babies diagnosed with infantile HPP had died within their first year.5,6
Childhood HPP is often first diagnosed by the general dental practitioner. It typically presents with early loss of the primary dentition (typically incisors) with intact roots, rickets, short stature, delayed walking or a waddling gait. These children may also have bone pain and delayed motor development. Symptoms may be very mild with only early tooth loss suggesting the underlying condition.
Adult HPP tends to manifest in middle age, but there is often a history of premature loss of primary teeth. Loss of the permanent teeth may be seen along with osteomalacia, osteoarthropathy and stress fractures. Delayed fracture healing is also seen and there is a high risk of non-union of the fractures. These patients have significant problems with the failure of implants such as hip and knee replacements, however, there is a case report of successful osteo-integration with dental implants.7
Adult HPP may be misdiagnosed as primary osteoporosis, and patients are often prescribed bisphosphonates to treat this. There is no evidence that such bisphosphonates are effective for the treatment of HPP and there is some evidence that it may in fact be deleterious in the condition.3
Odonto-hypophosphatasia is likely the commonest form of HPP.8 It is characterised solely by dental manifestations and is not associated with skeletal abnormalities.8,9,10 However, it has been noted that children with odonto-hypophosphatasia are on average, slightly shorter than their healthy counterparts. It is uncertain whether this reflects a minor skeletal impact.8
As with other forms of HPP the primary incisors are most likely to exfoliate early. Radiographs show enlarged pulp chambers and loss of alveolar bone height. Cases have been reported where initial blood ALP levels have been normal, even after repeated testing, and the diagnosis has only been confirmed after histological analysis of the exfoliated teeth.
A subset of patients diagnosed with odonto-hypophosphatasia will go on to develop Adult HPP in middle age with chronic joint pain and stress fractures. It is important when a child is diagnosed with suspected odonto-hypophosphatsia that a metabolic bone team are involved in the child's care to rule out any evidence of rickets or osteomalacia. These children and their families should also be warned of the potential to develop symptoms and it may be prudent to periodically review them to ensure they do not experience systemic complications in later life.11
Oral signs of HPP
Common dental signs:
-
Exfoliation of the primary dentition before the age of three
-
Exfoliation of primary teeth with roots intact
-
Alveolar bone loss
-
Loss of permanent teeth with no signs of periodontal inflammation
-
Enlarged pulp chambers and root canals
-
Thin dentinal walls.
Uncommon/poorly reported dental signs:
-
Delayed eruption of primary and permanent dentition
-
Small bulbous crowns
-
Enamel and dentine hypoplasia
-
Thin enamel that does not reach the cervical region
-
Cervical constrictions
-
Ankylosis of primary dentition
-
Rampant caries.12
While there are a range of differential diagnoses for the early loss of primary incisors (see Table 1), premature exfoliating of a primary incisor with intact roots and no signs of periodontal inflammation is virtually pathognomonic of HPP.
The more severe the phenotype, the more teeth are shed, with odonto-hypophosphatasia averaging four teeth lost before the age of five and mild childhood, severe childhood and infantile losing an average of six, seven and nine teeth respectively.8
Other dental features are likely to be secondary to the effects of HPP. For example, in her recent review paper Bloch-Zupan advises that the high rate of dental caries is likely to be due to poor appetite secondary to hypercalcaemia, which in turn results in a high sugar diet.4,10,11,13 The horizontal bone loss commonly seen in HPP is thought to be secondary to a deficiency of cementum and periodontal fibre attachment resulting in a lack of functional stimulus to the alveolus.14,15
It has also been shown that defects such as early loss of primary teeth due to lack of cementum are not necessary replicated in the permanent dentition and that defects such as thin dentine and enlarged pulp chambers may improve over time, with deposition of dentine reducing the size of pulp canals in adolescence.7,10,12
Histological signs
Histological analysis of exfoliated teeth is often required to confirm a diagnosis of odonto-hypophosphatasia, particularly when the alkaline phosphatase (ALP) levels are within a normal range.
The acellular cementum is almost completely absent. The cellular cementum may also be deficient. The collagenous fibrils of the periodontal ligament are not connected to the root via Sharpey's fibres. Instead the periodontal ligament and the exposed dentine are separated by a non-fibrillar layer.16 There is a reduction in periodontal ligament coverage, with plaque bacteria extending apically in areas of loss of periodontal ligament attachment.
Case series reports
Case 1 and 2
Case 1 and case 2 are siblings diagnosed with odonto-hypophosphatasia. They are unusual as neither of their parents is affected. They also have one unaffected sibling. Neither of the affected siblings display any systemic symptoms of HPP.
Case 1 (LMH)
Dental history and assessment
LMH presented to University Dental Hospital Manchester (UDHM) aged two, with early exfoliation of two lower primary incisors (71, 81). These had exfoliated before the child was 11 months old.
Radiographs from her initial assessment revealed bulbous crowns with large pulp chambers and vertical bone loss around the incisors. There is some mild bone loss around the primary molars (Figs 1 and 2).

Case 1 – Age 2 – Right and left lateral oblique radiographs showing large pulp chambers of the primary molars with mild bone loss

Case 1 – Age 2 – Lower anterior occlusal radiographs showing bone loss around the primary lateral incisors
Medical history
Nil relevant.
Family history
No family history of premature tooth loss or bone disease
Management and follow up
The child was referred to the Paediatric Department of the Royal Manchester Children's' Hospital (RMCH) where she was diagnosed with vitamin D deficiency. Blood tests taken at the time did, however, reveal low ALP levels.
She remained under the care of the Child Dental Health Department. Radiographs taken two years later show progressive bone loss around the primary molars and severe vertical bone loss around the 51 and 61 (Figs 3 and 4). At this stage she had lost all four lower primary incisors and much of her remaining primary dentition was mobile (Fig. 5).

Case 1 – Age 4 – Right and left lateral oblique radiographs showing progressive bone loss around the primary molars

Case 1 – Age 4 – Upper and lower anterior occlusal showing severe vertical bone loss around the 51 and 61 and early exfoliation of the 71, 72, 81and 82

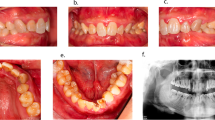
Case 1 – Age 4 – Clinical appearance of anterior dentition showing absent 72,71,82,81 and gingival recession on 52, 62, 73, 83
She was referred to the Metabolic Bone Diseases Team in RMCH when she was four years old and diagnosed with odonto-hypophosphatasia. Repeat blood tests revealed low ALP levels and slightly elevated phosphate.
She is under regular review with both the Metabolic Bone Diseases Team and the Child Dental Health Unit.
Case 2 (PH)
Dental history
LMH's younger sister PH, was referred to the Child Dental Health Department of the UDHM aged two.
She was missing seven primary incisors (52, 51, 61, 72, 71, 81, 82). The 51 was lost following a minor trauma at nine months of age. The 52, 61, 71 and 81 were lost less than three months later. Her parents had no memory of the lower lateral primary incisors exfoliating, so it is possible that these were congenitally absent, or the family did not notice their loss (Fig. 6).

Case 2 – Age 2 – Clinical appearance at presentation with loss of 52, 51, 61, 72, 71, 81 and 82
The patient was unable to tolerate anterior radiographs but right and left lateral oblique radiographs revealed bulbous crowns, large pulp chambers and mild to moderate bone loss around the primary molars (Fig. 7).

Case 2 – Age 2 – Left and right lateral oblique radiographs showing large pulp chambers of the primary molars with mild to moderate bone loss
Medical history
Nil relevant.
Family history
PH's elder sibling had been diagnosed with odonto-hypophosphatasia and had premature exfoliation of primary incisors.
Management and follow up
The child was referred to the Metabolic Bone Diseases Team in the RMCH, who confirmed the diagnosis of odonto-hypophosphatasia.
She is now under regular review with the Metabolic Bone team and the Child Dental Health Department.
Case 3 (NB)
Dental history
NB presented aged two to the Child dental Health Department of the UDHM, with missing 51, 61, 71, 81 and 82 (Fig. 8). His 61 was Grade I mobile. His mother gave a history of painless exfoliation of the 71 and 81 around the age of 20 months. The 51 and 61 were lost following a relatively minor trauma. The patient's mother felt that the 82 had never erupted, so this may have been congenitally absent.

Case 3 – Age 2 – Clinical appearance at presentation with loss of 51, 61, 71, 81 and 82
Medical history
The patient had been referred to a podiatrist by his GP due bowed legs.
Family history
Nil relevant.
Management and follow up
The patient was referred to the Metabolic Bone Disease team in the RMCH. Oral hygiene and diet advice were given. He was kept under regular review with the Child Dental Health Unit in UDHM and went on to lose the 51 and 61.
The patient was seen by a consultant in paediatric bone disease. His ALP levels were at the low end of normal and he had raised plasma pyridoxal-5-phosphate. It was also noted that he walked with an in-toeing gait and had some frontal bossing. He was diagnosed with likely odonto-hypophosphatasia, although there were some signs of childhood HPP. He will continue to be reviewed regularly by the Metabolic Bone Team. He is also under regular review with the Child Dental Health Unit.
Case 4 (TB)
Dental history
A four-year-old boy (TB) was seen by the Betsi Cadwaladr North Wales Community Dental Services as his mother was concerned by his mobile upper primary incisors (51, 61). He had previously lost his lower primary central incisors (71, 81) when he was two years old. The family had assumed that there had been some trauma but could not identify a specific incident at the time.
At the time of assessment, the primary upper central incisors were splayed and grade II mobile (Fig. 9). The remaining dentition was sound and there was no evidence of periodontal inflammation. An upper anterior occlusal radiograph showed significant bone loss around the 51 and 61 with no evidence of root resorption.

Case 4 – Age 4 – Upper anterior occlusal radiograph shows vertical bone loss and splayed roots
Medical history
TB had been under review by a paediatrician due to bowing of the legs and had annual radiographs of his legs until the previous year when he had been discharged. There was no other relevant medical history.
Family history
There was no family history of premature tooth loss or bone disease.
Treatment
The patient was seen by a consultant in paediatric dentistry, who then referred him for bone biochemistry blood tests. The samples revealed very low ALP levels, with all other results normal.
Management and follow up
The 61 was shed initially (Fig. 10) followed by 51, both with roots intact. An OPG revealed large bulbous crowns and mild bone loss around the primary molars (Fig. 11).

Case 4 – Age 4 – Clinical appearance with loss of 61, 71 and 81 – 61 exfoliated shortly after the initial assessment and radiographs

Case 4 – Age 4 – OPG radiograph showing generalised bone loss
The boy was referred to a community paediatrician for further input. A provisional diagnosis of childhood HPP has been made, pending further input from the paediatric medical team.
The child will remain under regular review with the community dental services.
Discussion
Estimating the incidence of HPP is difficult as the severe forms of the disease are very rare, and the mild forms often go undiagnosed. A recent molecular-based estimation of the incidence of HPP in the European population puts the incidence of severe forms at about 1:300,000 and mild forms at 1:6,370.17 However, studies from other populations estimate the incidence at 1:100,000 and 2–3:100,000.18,19
HPP is inherited via autosomal dominant and recessive mechanisms. The severe forms of the disease show a predominantly recessive mode of inheritance. The milder phenotypes show both dominant and recessive inheritance.
Genetic counselling is complicated by the fact that dominant forms have variable levels of clinical expression, so while a patient may test positive for low levels of ALP and carry the mutated TNSALP gene, they may not show any clinical signs of the disease.20,21
There are also cases where severely affected children have presented with compound heterozygous mutations, meaning that they have inherited two separate mutations, one from each parent who had not been previously diagnosed as having HPP. Often when questioned, the parents will recall symptoms from their own childhood which suggest milder forms of HPP, such as early loss of primary incisors, childhood rickets or joint pain.22
HPP is caused by mutations in the ALPL gene on chromosome 1p36.1, which encodes the enzyme TNSALP, resulting in decreased levels of enzyme activity and increased levels of its substrates.21 So far more than 300 different mutations have been recorded in the ALPL gene mutations database (http://www.sesep.uvsq.fr/03_hypo_mutations.php). Most these are missense mutations which may explain the wide variation of clinical presentation of HPP.22
TNSALP is found throughout the body, but the full extent of its functions has not been elucidated. It is found in high concentration in the liver, kidneys and bones, enamel, cementum, dentine and alveolar bone.23
TNSALP cleaves extracellular pyridoxal-5'-phosphate (PLP or vitamin B6), phosphoethanolamine (PEA) and inorganic pyrophosphates (PPi). Its effects on PPi (inorganic pyrophosphates) are considered the most significant aspects in explaining the skeletal and dental effects of HPP.16,24
Inorganic pyrophosphate (PPi) suppresses the growth of hydroxyapatite crystals, and therefore inhibits calcification of skeletal and dental tissues. In normal individuals, TNSALP inactivates PPi. Patients with HPP have a deficiency or defect in TNSALP resulting in high levels of PPi and an inhibition of skeletal and dental mineralisation. The degree to which skeletal mineralisation is affected depends on the level of severity of the phenotype.
TNSALP also plays an important role in vitamin B6 metabolism. The TNSALP enzyme is required to dephosphorylate the active metabolite of vitamin B6 (PLP) for it to cross the blood brain barrier and for cellular uptake.25 PLP appears to be essential for the synthesis of neurotransmitters such as GABA, 5-hydroxytryptamine and dopamine. Thus, a lack of TNSALP may affect the excitation and inhibition of the neural networks, resulting in the vitamin B6 responsive seizures seen in the severe forms of the disease.3,26 Research is ongoing regarding the full neurological effects of TNSALP, but it thought that TNSALP is involved in fundamental processes in brain development such as proliferation and differentiation of neural stem cells, myelination, axonal growth and the maturation of synapses and their maintenance. Studies in mice have suggested that TNSALP is involved in pain control. Bone pain has been reported in a number of clinical cases of mild hypophosphatasia in adolescents and adults, and would be a particularly relevant side effect for dental practitioners.27,28 Patients with HPP are also at increased risk of depression and anxiety and it has been suggested that this may be related to low circulating levels of TNSALP, although the mechanism for this is still unknown.3
Diagnosis and treatment of HPP
The diagnosis of HPP is based on clinical symptoms, biochemistry (ALP level) and genetic testing. Repeated bloods are needed to show consistently low ALP levels. Normal levels for ALP vary with age and sex so it is important that the correct scale is used to compare results. Increased urinary levels of phosphoethanolamine (PEP) are also used as a diagnostic tool, but this is less specific. Low levels of ALP in conjunction with clinical signs can be diagnostic for HPP, however, genetic analysis is becoming an ever more important tool in the diagnosis of HPP, particularly for the more moderate forms, as well as in odontohypophosphatasia, where clinical and biochemical signs may not be as clear cut.29
Until very recently, treatment for all types of HPP was largely supportive.
Perinatal, infantile and severe childhood HPP had a survival rate of 50%. Respiratory distress was treated with ventilation, a low calcium diet was implemented to reduce the risk of nephrocalcinosis and vitamin B6 supplementation was used to treat vitamin B6 deficient seizures, although these seizures often indicated that the disease would be fatal. Surgery was required to correct craniosynostosis.
These treatments are still used, but they are now being supplemented by enzyme replacement therapy (tradename Asofatase Alfa) which is showing astonishing results in the remineralisation of the skeletons of children with perinatal, infantile and childhood HPP, with 95% of infants surviving their first year.30 It is too early to tell whether enzyme replacement therapy will also improve the dental outcomes for these patients; however, lab tests of the same drug in mice suggested that there was a reduction in enamel and dentine defects following administration of the drug, but acellular cementum and alveolar bone were still affected and the incisors were still lost.31
Enzyme replacement therapy has until very recently only been approved for treatment of perinatal and infantile forms of the disease in the UK and Europe. However, as of July 2017 NICE and the manufacturers Alexion announced that an agreement has been reached that will mean that the drug will become available to patients with childhood onset HPP, on a 5-year managed access agreement.32 This will broaden the availability of the drug to children and adults who are most severely affected by the disease. These patients will be followed and monitored for treatment outcomes and this will provide invaluable information on the effect of treatment on all aspects of HPP.
There is also a world-wide registry of patients with HPP which has been collecting data since December 2014 (https://clinicaltrials.gov/ct2/show/NCT02306720). Any patient with a confirmed diagnosis of HPP, who is not undergoing treatment with enzyme replacement therapy, may be included on the database with the aim of providing a longitudinal, real life clinical profile of patients with HPP. This is particularly important to provide an accurate clinical and epidemiological picture of the underreported and milder forms of HPP, such as odontohypophosphatasia. There are a number of centres in the UK which are participating in this trial.
Regular dental monitoring and good oral hygiene are still essential for all patients with HPP. An accurate diagnosis of their condition is also important to avoid potentially deleterious treatments such as bisphosphonates and calcium supplementation.3
Conclusion
HPP is classified as a rare disease. However, particularly with current treatment advances, GDPs may well encounter patients with this disease over the course of their working lives.
Patients with severe forms of HPP, who would previously have died in infancy, are now surviving thanks to recently available enzyme therapy and the initial trial subjects are entering early childhood. These will come under the care of primary and secondary care clinicians in the coming years and all GDPs should be aware of HPP in order to manage these children appropriately.
Most importantly, dentists in primary care are critical in the detection and diagnosis of both childhood and adult forms of hypophosphatasia. A GDP may save their patient years of pain and mismanagement by linking their medical history of osteopenia and fractures with a history of early tooth loss or by referring a child with non-traumatic early tooth loss on to a specialist centre.
As enzyme replacement therapy is becoming more widely available for all ages, a vigilant GDP who makes an early diagnosis is the key to the prevention of a lifetime of bony degeneration, pain and tooth loss.
Glossary
Phenotype – clinical presentation or symptoms of disease
Genotype – the particular type and arrangement of genes that each organism has
Missense mutation – these are point mutations in a single nucleotide in the DNA sequence which causes a change in one amino acid in the protein being encoded. This change of amino acid may or may not affect the protein's function within the body
Rachitic chest deformity – pulled in sternum or 'pigeon breast', pronounced 'knobs' at the costo-chondrial junction of the ribs and a flaring of the ribs over the diaphragm
Craniosynostosis – premature fusion of the cranial plates
Nephrocalcinosis – deposition of calcium-phosphate (mineralised nodules) within the kidneys tissues and can eventually lead to renal failure
Osteomalacia – adult rickets or soft bones
Osteoarthropathy – bone and joint pain
References
Bishop N . Clinical management of hypophosphatasia. Clin Cases Miner Bone Metab 2015; 12: 170–173.
Whyte M P, Greenberg C R, Salman N J et al. Enzyme-replacement therapy in life-threatening hypophosphatasia. N Engl J Med 2012; 366: 904–913.
Hofmann C, Girschick H J, Mentrup B et al. Clinical Aspects of Hypophosphatasia: An Update. Clin Rev Bone Min Metabol 2013; 11: 60–70.
Bloch-Zupan A . Hypophosphatasia: diagnosis and clinical signs – a dental surgeon perspective. Int J Paediatr Dent 2016; 26: 426–438.
Whyte M, Leung E, Wilcox W et al. Hypophosphatasia: a retrospective natural history study of the severe perinatal and infantile forms. Bone Abstracts 2014; 3: 364.
Leung E C, Mhanni A A, Reed M, Whyte M P, Landy H, Greenberg C R . Outcome of perinatal hypophosphatasia in manitoba mennonites: a retrospective cohort analysis. JIMD Reports 2013; 11: 73–78.
Lynch C D, Ziada H M, Buckley LA, O'Sullivan V R, Aherne T, Aherne S . Prosthodontic rehabilitation of hypophosphatasia using dental implants: a review of the literature and two case reports. J Oral Rehabil 2009; 36: 462–468.
Whyte M P, Zhang F, Wenkert D et al. Hypophosphatasia: validation and expansion of the clinical nosology for children from 25 years experience with 173 paediatric patients. Bone 2015; 75: 229–239.
Rockman-Greenberg C . Hypophosphatasia. Pediatr Endocrinol Rev 2013; 10 Suppl 2: 380–388.
Reibel A, Maniere M C, Clauss F et al. Orodental phenotype and genotype findings in all subtypes of hypophosphatasia. Orphanet J Rare Dis 2009; 4: 6.
Mori M, DeArmey S L, Weber T J, Kishnani P S . Case series: Odontohypophosphatasia or missed diagnosis of childhood/adult-onset hypophosphatasia? – Call for a long-term follow-up of premature loss of primary teeth. Bone Reports 2016; 5: 228–232.
Chapple I L . Hypophosphatasia: dental aspects and mode of inheritance. J Clin Periodontol 1993; 20: 615–622.
Beck C, Morbach H, Richl P, Stenzel M, Girschick H J . How can calcium pyrophosphate crystals induce inflammation in hypophosphatasia or chronic inflammatory joint diseases? Rheumatol Int 2009; 29: 229–238.
Beumer J, 3rd, Trowbridge H O, Silverman S, Jr ., Eisenberg E . Childhood hypophosphatasia and the premature loss of teeth. A clinical and laboratory study of seven cases. Oral Surg Oral Med Oral Pathol 1973; 35: 631–640.
Foster B L, Ramnitz M S, Gafni R I et al. Rare bone diseases and their dental, oral, and craniofacial manifestations. J Dent Res 2014; 93 (7 Suppl): 7S–19S.
van den Bos T, Handoko G, Niehof A et al. Cementum and Dentin in Hypophosphatasia. J Dent Res 2005; 84: 1021–1025.
Mornet E, Yvard A, Taillandier A, Fauvert D, Simon-Bouy B . A molecular-based estimation of the prevalence of hypophosphatasia in the European population. Ann Hum Genet 2011; 75: 439–445.
Taketani T, Onigata K, Kobayashi H, Mushimoto Y, Fukuda S, Yamaguchi S . Clinical and genetic aspects of hypophosphatasia in Japanese patients. Arch Dis Child 2014; 99: 211–215.
Fraser D . Hypophosphatasia. Am J Med 1957; 22: 730–746.
Mornet E . Hypophosphatasia. Orphanet J Rare Dis 2007; 2: 40.
Whyte M P . Hypophosphatasia and the role of alkaline phosphatase in skeletal mineralization. Endocr Rev 1994; 15: 439–461.
Hofmann C, Girschick H, Mornet E, Schneider D, Jakob F, Mentrup B . Unexpected high intrafamilial phenotypic variability observed in hypophosphatasia. Eur J Hum Genet 2014; 22: 1160–1164.
Liu H, Li J, Lei H, Zhu T, Gan Y, Ge L . Genetic aetiology and dental pulp cell deficiency of hypophosphatasia. J Dent Res 2010; 89: 1373–1377.
Foster B L, Nagatomo K J, Nociti F H, Jr . et al. Central role of pyrophosphate in acellular cementum formation. PloS One 2012; 7: e38393.
Cole D E . Hypophosphatasia update: recent advances in diagnosis and treatment. Clin Genet 2008; 73: 232–235.
Demirbilek H, Alanay Y, Alikasifoglu A et al. Hypophosphatasia presenting with pyridoxine-responsive seizures, hypercalcaemia, and pseudotumour cerebri: case report. J Clin Res Pediatr Endocrinol 2012; 4: 34–38.
Fonta C, Barone P, Rodriguez Martinez L, Negyessy L . Rediscovering TNAP in the Brain: A Major Role in Regulating the Function and Development of the Cerebral Cortex. Subcell Biochem 2015; 76: 85–106.
Whyte M P, Wenkert D, McAlister W H et al. Chronic recurrent multifocal osteomyelitis mimicked in childhood hypophosphatasia. J Bone Miner Res 2009; 24: 1493–1505.
Hollis A, Arundel P, High A, Balmer R . Current concepts in hypophosphatasia: case report and literature review. Int J Paediatr Dent 2013; 23: 153–159.
Whyte M P, Rockman-Greenberg C, Ozono K et al. Asfotase Alfa Treatment Improves Survival for Perinatal and Infantile Hypophosphatasia. J Clin Endocrinol Metab 2016; 101: 334–342.
Gasque K C, Foster B L, Kuss P et al. Improvement of the skeletal and dental hypophosphatasia phenotype in Alpl/mice by administration of soluble (non-targeted) chimeric alkaline phosphatase. Bone 2015; 72: 137–147.
NICE. New Stensiq deal paves way for NICE approval of life saving drug. 2017. Available at https://www.nice.org.uk/news/article/stensiq-new-deal-paves-way-for-nice-approval-of-life-saving-drug (accessed May 2018).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Feeney, C., Stanford, N., Lee, S. et al. Hypophosphatasia and the importance of the general dental practitioner – a case series and discussion of upcoming treatments. Br Dent J 224, 937–943 (2018). https://doi.org/10.1038/sj.bdj.2018.441
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bdj.2018.441
This article is cited by
-
The diagnosis of hypophosphatasia in children as a multidisciplinary effort: an expert opinion
Journal of Endocrinological Investigation (2023)
-
The impact of enzyme replacement therapy on the oral health manifestations of hypophosphatasia among children: a scoping review
European Archives of Paediatric Dentistry (2023)
-
Evaluation of alveolar bone hypomineralization in pediatric hypophosphatasia using orthopantomography
Scientific Reports (2022)
-
Effects of Infantile Hypophosphatasia on Human Dental Tissue
Calcified Tissue International (2022)
-
Dental effects of enzyme replacement therapy in case of childhood-type hypophosphatasia
BMC Oral Health (2021)
