
Abstract
We present an extensive and diverse database of peptide conformational energies. Our database contains five different classes of model geometries: dipeptides, tripeptides, and disulfide-bridged, bioactive, and cyclic peptides. In total, the database consists of 3775 conformational energy data points and 4530 conformer geometries. All the reference energies have been calculated at the LC-ωPBE-XDM/aug-cc-pVTZ level of theory, which is shown to yield conformational energies with an accuracy in the order of tenths of a kcal/mol when compared to complete-basis-set coupled-cluster reference data. The peptide conformational data set (PEPCONF) is presented as a high-quality reference set for the development and benchmarking of molecular-mechanics and semi-empirical electronic structure methods, which are the most commonly used techniques in the modeling of medium to large proteins.
Design Type(s) | modeling and simulation objective • chemical structure modeling objective • molecular structure analysis objective • data integration objective |
Measurement Type(s) | energy |
Technology Type(s) | computational modeling technique |
Factor Type(s) | class |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others



Background & Summary
The structure and function of proteins are governed by the intermolecular interactions between their building blocks, amino acids. The accurate prediction of protein folding and ligand binding energetics depends on how well the computational modeling method employed captures the interactions between individual amino acids. For this reason, results obtained from the computational methods commonly employed to model proteins, such as force field and semi-empirical electronic structure methods, are usually compared to, and parametrized against, those obtained from higher-level computational methods. A database of peptide conformational energies is an ideal benchmark set for testing and parameterizing computational methods since conformational energies capture the interplay between bonded and non-bonded interactions that are present in proteins.
Similar sets to the one proposed in this work are available in the literature, but they tend to be small and focus on specific peptide interactions or otherwise focus exclusively on single amino acids. In 2008, Hobza and co-workers presented a benchmark database of conformational energies for a set of 76 conformers of four tripeptides and a dipeptide containing aromatic side chains1. The conformational energies were calculated at the CCSD(T)/complete-basis-set (CBS) level of theory and, in the same work, were used to assess lower-level quantum-mechanical (QM) methods. The reference data for a subset of Hobza’s set (named PCONF) was updated by Smith and co-workers2, and later by Goerigk and co-workers3. Wilke et al. proposed a set of conformational energies for cysteine known as CYCONF4, eight conformational energies of tetrapeptide conformers were proposed by Goerigk et al.5, and Ropo et al. presented a conformer data set of capped and uncapped versions of proteinogenic amino acids and their interactions with divalent cations evaluated at ‘PBE + vdW’ level of theory6. More recently, Martin and co-workers re-optimized the conformer structures of twenty proteinogenic amino acids from a previously published set by Yuan, Mills, Popelier, and Jensen (the YMPJ database)7,8. These structures were then used to generate a new conformational energy database of isolated amino acid monomers containing 466 data points. A database of macrocyclic conformers, called MPCONF196, has recently been published9. The MPCONF196 set contains conformational energies of eight macrocyclic compounds including cyclic peptides of varying sizes. To our knowledge, MPCONF196 is the only set in the literature that considers cyclic peptides. Several of the data sets described above have been compiled into supersets. Hobza’s 2008 data set was included as a subset of the MPCONF196 benchmark database1,9. Similarly, the CYCONF, PCONF, TPCONF, and YMPJ sets of conformational energies were incorporated in the GMTKN databases by Grimme and co-workers3,10,11.
To best of our knowledge, an extensive database of polypeptide conformations is not yet available in the literature. It is likely that the absence of a comprehensive data set rests on the fact that structural complexity and the computational cost of obtaining reference-quality data increases with system size. A comprehensive set of data that contains reference conformational energies on a diversity of small peptides would provide valuable information to those engaged in the development of atomistic computational methods for protein modeling. Producing such a database of conformational energies of diverse polypeptides would ensure a uniform high-quality standard in the reference data by eliminating the need to collect and verify data gathered from various sources, which may differ substantially in their mode of generation and quality.
In this work, we have undertaken a substantial computational effort to generate a large, comprehensive polypeptide conformational energy data set using dispersion-corrected range-separated density-functional theory. The data set has several important features: 1) The conformational energies were obtained using a single computational method, which results in data with uniform quality; 2) The quality of the results obtained from the computational method we used to obtain the conformational energies is benchmarked against those obtained using complete-basis-set coupled-cluster methods. This provides a means for assessing the quality of our database; 3) The computational method we used to obtain conformational energies is of much higher quality than conventional force field methods used for large-scale protein modeling and is therefore fit for testing and parametrization of conventional force field methods. Therefore, our data can be used for molecular mechanics force field development12–14, and parametrization of cost-effective computational procedures like Atom-Centered Potentials (ACP)15,16 and other low-cost correction approaches17–19. It also serves as a direct source for comparative benchmark studies of various energy functions20–27, semi-empirical approaches28–40, and inexpensive electronic structure methods41–47 in the context of protein modeling.
Methods
Generation of the model geometries
The PEPCONF set comprises five different kinds of model systems:
-
Dipeptides: All unique pairs of the twenty standard proteinogenic amino acids were selected (for instance, ALA-GLY and GLY-ALA were considered to be the same from the perspective of side chain-side chain interactions), leading to 136 neutral and 74 charged dipeptide geometries.
-
Tripeptides: Unique combinations of tripeptide sequences were selected similarly but, in order to limit the number of combinations, one representative amino acid was chosen from each of the side-chain categories in Fig. 1a: Leucine for aliphatic, Proline for cyclic, Tryptophan for aromatic, Tyrosine for hydroxylic, Methionine for sulfur-containing, neutral Glutamic acid for acidic, Histidine for basic, and Glutamine for amidic side-chains. This yielded a total of 288 unique combinations of amino acid trimers.
Figure 1: Molecular structure of the amino acids and representative peptide model systems considered in this work. 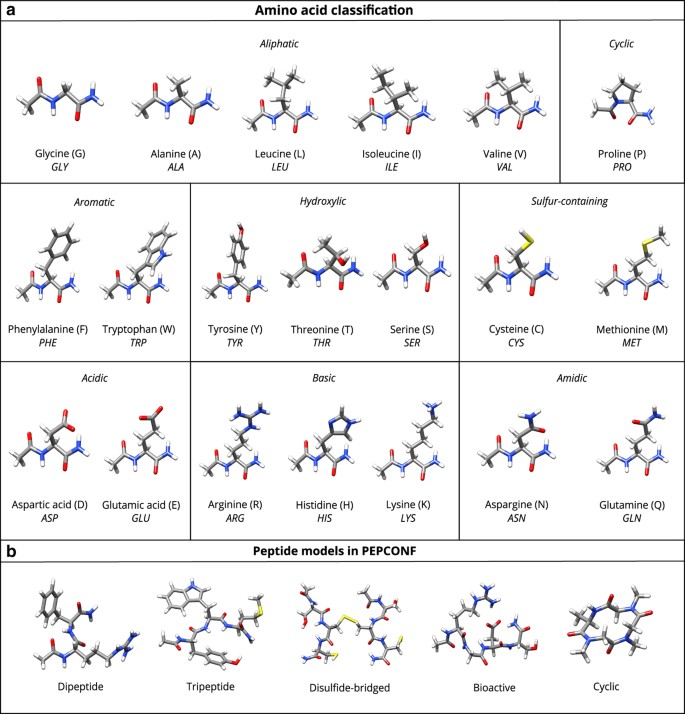
(a) The classification of the twenty standard proteinogenic amino acids by the nature of their side-chains. The N-terminal and C-terminal are capped with acetyl and primary amide group, respectively. The single- and three-letter codes for each amino acid are also provided. (b) A representative candidate from each of the five different classes of peptide model systems considered in the PEPCONF data set.
-
Disulfide-bridged: Oligopeptides where the two cysteine residues are internally connected via a disulfide bond (154 model systems).
-
Bioactive: Oligopeptides where the chosen residue sequences were found to be associated with bio-functionality as reported in the literature48 (39 model systems).
-
Cyclic: Oligopeptides where the N-terminus and C-terminus of the peptide backbone are connected to form a circular bond (64 model systems).

Structures
The initial gas-phase model geometries of the dipeptides, tripeptides, and bioactive peptides were generated using the sequence command in the tleap tool of Amber16 software package49–51. The disulfide-bridged and cyclic peptides were generated manually from structures taken from the Protein Data Bank (PDB)52,53 and the Cambridge Structural Database (CSD)54,55, respectively. The N-terminal(s) and C-terminal(s) of all the representative model structures except for cyclic peptides were capped with acetyl (ACE) and primary amide (NHE) groups, respectively. The complete list of all the peptide structures considered in this work is provided in the supplementary file accompanying this article (Supplementary File 1).
The initial model geometries of disulfide-bridged oligopeptides were generated using an in-house fragmentation code and a combination of various Amber16 tools like pdb4amber, tleap, and pytleap. Representative structures were initially obtained from searches of the Protein Data Bank (PDB) using the online advanced search interface with the following criteria: (i) only one disulfide bond, (ii) X-ray resolution between 2.5–3.5 Å, (iii) no modified polymeric residues, (iv) no free ligands, and (v) representative structures at 100% sequence identity. The resulting 191 hits were then processed with the pdb4amber tool to remove the water molecules from the PDB files and to select the most populous conformer. We then discarded 37 out of the 191 clean PDB files because the most populated conformer did not contain a disulfide bond. Finally, the clean PDB files were truncated using our fragmentation code and the disulfide-bridged cysteine residues of each model system were extracted along with at most four neighboring backbone residues. Each system was manually checked and then processed with pytleap and tleap to add the missing hydrogen atoms and terminal capping groups.
The initial model geometries of cyclic peptides were found using the Conquest software package to search for crystal structures in the Cambridge Structural Database. Cyclic sequences of proteinogenic amino acids were searched using the peptide building query tool. The following search criteria were used: (i) 3D coordinates must have been determined, (ii) R-factor less than or equal to 0.05, (iii) only non-disordered crystals, (iv) no errors present, (v) no ions present. The resulting structures were then exported to ‘mol2’ files which were converted to ‘xyz’ format using Openbabel56,57 and loaded in the Avogadro58,59 software package for visual inspection. Structures without a proper cyclic peptide backbone were not considered. Finally, the missing H-atoms were added using Avogadro.
The initial geometries of all the model systems, with the exception of cyclic peptides, were subjected to Amber ff14SB21 unconstrained force field energy relaxations using the sander module of Amber16.
Conformational search
A force field-based high-temperature molecular dynamics (HTMD) simulation approach60 was used in a manner similar to previous studies in the literature61–64 to generate the conformers for the non-cyclic peptides. Initial structures were subjected to canonical ensemble simulations with Langevin dynamics scaling at a temperature of 900 K. The MD steps were performed with the sander module of Amber16 without solvent or periodicity. A heating (equilibration) step of 200 picoseconds was followed by a production run of 4.2 nanoseconds. Structures along the trajectory of the production run were sampled at uniform time intervals, resulting in 4000 conformers for each peptide model system. Each conformer was subjected to energy minimization using the Amber ff14SB force field.
The Amber ff14SB force field does not contain parameter for cyclic peptides. We therefore used the RDKit software package65 to generate cyclic peptide conformations. The accuracy and speed of RDKit’s conformer generation approach in comparison to other freely available conformer generation toolkits was reviewed in ref. 66, where it was reported that the program is suitable for less flexible molecules like the cyclic peptides considered in this work. A distance-geometry-based stochastic method67 was used to yield 100 conformers for each cyclic peptide. A very similar approach was recently used to generate the 3D conformations reported in the ANI-1 data set68.
Conformer binning strategy
The list of relaxed conformers was pruned using a binning strategy. Each set of non-cyclic conformers was sorted according to the force field energy, from most to least stable. The least stable conformers from the upper half of the list were removed, and the remainder of the list was divided into thirty equal energy intervals. From each interval, one conformer geometry was selected and was subjected to a single-point energy calculation with the BLYP gradient-corrected density functional69,70, and the 6-31 G∗ basis set71,72, combined with Grimme’s D3 dispersion-correction method73,74 with Becke-Johnson (BJ) damping function75–81 and recently developed basis set incompleteness potentials (BSIP)82. The calculations with the BLYP-D3(BJ)/6-31 G∗-BSIP level of theory were carried out using the Gaussian software package83,84, with SCF convergence criterion of 10−6 Hartrees and pruned integration grid with 99 radial and 590 angular points (ultrafine grid). The resulting BLYP-D3(BJ)/6-31 G∗-BSIP energies were used to select the six most stable conformers out of the thirty for entry into the PEPCONF data set.
In the case of the cyclic peptides, the 100 conformers generated by RDKit were geometry-optimized at the BLYP-D3(BJ)/MINIs-BSIP69,70,73–82,85 level of theory using the Gaussian package. The calculations employed SCF convergence criterion of 10−8 Hartrees, ultrafine integration grid, and the default optimization convergence criteria (maximum force = 4.5 × 10−4 Hartrees/Bohr, RMS force = 3 × 10−4 Hartrees/Bohr, maximum displacement = 1.8 × 10−3 Bohr, RMS displacement = 1.2 × 10−3 Bohr). The equilibrium geometries were sorted by energy and six conformations from equally-spaced energy intervals covering the whole energy range were then selected. The six conformations were then subjected to further geometry optimizations using BLYP-D3(BJ)/6-31 G∗-BSIP with the same SCF and grid settings as above and a ‘verytight’ optimization convergence criteria (maximum force = 2 × 10−6 Hartrees/Bohr, RMS force = 1 × 10−6 Hartrees/Bohr, maximum displacement = 6 × 10−6 Bohr, RMS displacement = 4 × 10−6 Bohr).
Generation of the reference energies
The PEPCONF data set contains 5 relative conformational energies (from the 6 conformations) for each peptide model system considered, yielding a total of 3775 data points and 4530 conformer structures. The reference energies were calculated with the LC-ωPBE86,87 range-separated density functional, and the aug-cc-pVTZ basis set of Dunning and co-workers88–90, combined with the exchange-hole dipole moment (XDM) dispersion-correction technique75–81. The rationale for this choice is that it offers a good compromise between accuracy and speed, and we expect range-separated hybrid functionals to minimize the impact of functional delocalization error on zwitterionic and charged species91. The resulting DFT-based approach was chosen as the reference level because of its excellent performance for gas-phase results of relative conformational energies (see Technical Validation).
A wave-function based approach like the “gold-standard” CCSD(T)/CBS would provide more reliable relative conformer energies92,93. However, CCSD(T)/CBS calculations are not feasible for the quite large systems (23–166 number of atoms) included in the data set. In addition, the PEPCONF data set is intended as a database for parametrization and benchmarking of force fields, semi-empirical methods and other low computational cost methods, which have much higher errors in conformational energies than those associated with LC-ωPBE-XDM/aug-cc-pVTZ. Future revisions of the PEPCONF set may become possible as computing power increases and approximate but accurate CCSD(T) methods are developed94,95.
Code availability
The molecular dynamics simulations were carried out using Amber16, which is available from http://ambermd.org/ through a commercial license. The Amber16 tools pdb4amber, tleap, and pytleap used for peptide structure editing and manipulation are part of the Amber16 software package. The Cambridge Structural Database 2018 and the Conquest program are distributed under a commercial license at https://www.ccdc.cam.ac.uk/. RDKit is an open-source cheminformatics software made available under the Berkeley Software Distribution (BSD) license at https://www.rdkit.org/. The OpenBabel software package was used for file-type interconversions and is freely available from http://openbabel.org/ under the GPL license. The Avogadro molecular editor and visualizer is an open-source program available at https://avogadro.cc/. The quantum-mechanical calculations were performed using the Gaussian09/16 software packages, which can be purchased from Gaussian Inc. (http://gaussian.com/) under a commercial license. Finally, the Basis-Set Incompleteness Potentials (BSIP) for BLYP-D3(BJ)/MINIs and BLYP-D3(BJ)/6-31 G∗ level of theory can be obtained from the Supporting Information of ref. 82.
Data Records
The conformational reference energies (in kcal/mol) and coordinates (in Å) of the conformer geometries present in the PEPCONF data set are publicly available free-of-charge from the Figshare (Data Citation 1) and GitHub (https://github.com/aoterodelaroza/pepconf) repositories in the plain-text DB-format described in Table 1. The atomic coordinates of the conformer geometries are also stored in a plain-text XYZ-format. The PEPCONF set contains five DB-format and six XYZ-format files for each peptide model system. In total, deposited files include 3775 DB-format files and 4530 XYZ-format files stored in their respective peptide classification directory named Dipeptide, Tripeptide, Disulfide, Bioactive, and Cyclic. A CSV-format file is also provided in each directory and contains the reference energy values for all the peptide systems in that directory.
File format
For each molecule, the reference conformational energy, relative to the lowest-energy structure, and the atomic coordinates are stored in a file named MoleculeName_A.db, where A is the conformer identification number (1–5, ordered from lowest to highest relative energy). The Cartesian coordinates of the atoms are stored in files named MoleculeName_B.xyz, where B is 0–5 (ordered from lowest to highest relative energy), with 0 representing the lowest-energy reference structure.
The DB-format file contains a header line specifying the reference energy value (in kcal/mol) followed by two ‘molc’ (short for molecule) blocks containing a unique integer identifier, charge, multiplicity, and the atomic coordinates (in Å) of the peptide conformer and its corresponding lowest energy conformer. The XYZ-format file contains a header line defining the number of atoms N, a comment line containing the charge and multiplicity, and N lines with each containing element type and X, Y, Z coordinates (in Å). The CSV-format file is a comma-separated plain-text file containing multiple lines and three columns. The columns are: (i) identification number, (ii) name of the peptide, and (iii) reference conformational energy (in kcal/mol).
Technical Validation
The LC-ωPBE-XDM/aug-cc-pVTZ method was chosen as the reference level of theory for the single-point energy calculations of all the conformers in the PEPCONF data set. To justify the use of LC-ωPBE-XDM/aug-cc-pVTZ as the reference level, we checked its performance on several benchmark sets for conformational energies from the literature. The performance of LC-ωPBE-XDM/aug-cc-pVTZ is quantified in terms of the mean absolute error (MAE) relative to higher-level reference data. For Hobza’s 2008 conformer database of small peptides1, the MAE of LC-ωPBE-XDM/aug-cc-pVTZ relative to the CCSD(T)/CBS reference energies is 0.52 kcal/mol. The LC-ωPBE-XDM/aug-cc-pVTZ method also yields an MAE of 0.48 kcal/mol for the YMPJ8 set of amino acid conformers relative to the MP2-F12/cc-pVTZ-F12 + [CCSD(Ts)-F12b – MP2-F12]/cc-pVDZ-F12 data. The MAE of LC-ωPBE-XDM/aug-cc-pVTZ for the smaller peptide conformer sets are: 0.62 kcal/mol for CYCONF4,11 (relative to CCSD(T)/CBS), 0.61 kcal/mol for PCONF2 (relative to CCSD(T∗∗)-F12a/CBS) and 0.60 kcal/mol for TPCONF3,5 (relative to CCSD(T)/CBS).
Although they do not involve peptides, there are several other sets that can be used to validate the performance of LC-ωPBE-XDM/aug-cc-pVTZ for its ability to predict conformer energies. For example: 0.12 kcal/mol for ACONF11,96 (n-alkane conformations, relative to W1h-val), 0.07 kcal/mol for BUT14DIOL97 (conformations of butane-1,4-diol, relative to CCSD(T)-F12b/cc-pVTZ-F12), 0.75 kcal/mol for CCONF98 (conformations of glucose and α-maltose, relative to DLPNO-CCSD(T)/CBS), 0.21 kcal/mol for MCONF99 (melatonin conformations, relative to CCSD(T)/CBS), 0.24 kcal/mol for SCONF11,100 (sugar conformations, relative to CCSD(T)/CBS), and 0.62 kcal/mol for UpU46101 (RNA backbone conformations, relative to DLPNO-CCSD(T)/CBS). For comparison with peptide based non-covalent interaction energy data sets, LC-ωPBE-XDM/aug-cc-pVTZ gives MAE of 0.33 and 0.23 kcal/mol relative to DW-CCSD(T)-F12/aug-cc-pV(D + d)z for the BBI102 and SSI102 sets of backbone-backbone and sidechain-sidechain interactions, respectively. LC-ωPBE-XDM/aug-cc-pVTZ also yields an MAE of 0.28 and 0.18 kcal/mol for the S22 and S66 sets and 0.23 and 0.15 kcal/mol for the S22x5 and S66x8 sets of non-covalent binding energies calculated at the CCSD(T)/CBS limit, respectively103–107. A detailed analysis of the LC-ωPBE-XDM/aug-cc-pVTZ method for non-covalent interactions and thermochemistry can also be found in ref. 108.
Additional information
How to cite this article: Prasad, V. K. et al. PEPCONF, a diverse data set of peptide conformational energies. Sci. Data. 6:180310 doi: 10.1038/sdata.2018.310 (2019).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
References
Valdés, H., Pluháčková, K., Pitonák, M., Řezáč, J. & Hobza, P. Benchmark database on isolated small peptides containing an aromatic side chain: comparison between wave function and density functional theory methods and empirical force field. Phys. Chem. Chem. Phys. 10, 2747–2757 (2008).
Smith, D. G. A., Burns, L. A., Patkowski, K. & Sherrill, C. D. Revised damping parameters for the D3 dispersion correction to density functional theory. J. Phys. Chem. Lett. 7, 2197–2203 (2016).
Goerigk, L. et al. A look at the density functional theory zoo with the advanced GMTKN55 database for general main group thermochemistry, kinetics and noncovalent interactions. Phys. Chem. Chem. Phys. 19, 32184–32215 (2017).
Wilke, J. J., Lind, M. C., Schaefer, H. F. III., Császár, A. G. & Allen, W. D. Conformers of gaseous cysteine. J. Chem. Theory Comput. 5, 1511–1523 (2009).
Goerigk, L., Karton, A., Martin, J. M. L. & Radom, L. Accurate quantum chemical energies for tetrapeptide conformations: why MP2 data with an insufficient basis set should be handled with caution. Phys. Chem. Chem. Phys. 15, 7028–7031 (2013).
Ropo, M., Schneider, M., Baldauf, C. & Blum, V. First-principles data set of 45,892 isolated and cation-coordinated conformers of 20 proteinogenic amino acids. Sci. Data 3, 160009 (2016).
Yuan, Y., Mills, M. J. L., Popelier, P. L. A. & Jensen, F. Comprehensive analysis of energy minima of the 20 natural amino acids. J. Phys. Chem. A 118, 7876–7891 (2014).
Kesharwani, M. K., Karton, A. & Martin, J. M. L. Benchmark ab initio conformational energies for the proteinogenic amino acids through explicitly correlated methods. Assessment of density functional methods. J. Chem. Theory Comput 12, 444–454 (2016).
Řezáč, J., Bím, D., Gutten, O. & Rulíšek, L. Toward accurate conformational energies of smaller peptides and medium-sized macrocycles: MPCONF196 benchmark energy data set. J. Chem. Theory Comput. 14, 1254–1266 (2018).
Goerigk, L. & Grimme, S. A general database for main group thermochemistry, kinetics, and noncovalent interactions−assessment of common and reparameterized (meta-)GGA density functionals. J. Chem. Theory Comput. 6, 107–126 (2010).
Goerigk, L. & Grimme, S. Efficient and accurate double-hybrid-meta-GGA density functionals − evaluation with the extended GMTKN30 database for general main group thermochemistry, kinetics, and noncovalent interactions. J. Chem. Theory Comput. 7, 291–309 (2011).
Sakae, Y. & Okamoto, Y. In Computational Methods to Study the Structure and Dynamics of Biomolecules and Biomolecular Processes: From Bioinformatics to Molecular Quantum Mechanics. Springer Series in Bio-/Neuroinformatics Vol. 1, ed. Liwo A. Ch. 7 (Springer, Berlin: Heidelberg, 2014).
Lopes, P. E. M., Guvench, O. & MacKerell, A. D. Jr. In Molecular Modeling of Protiens. 2nd edn. Methods in Molecular Biology (Methods and Protocols) Vol. 1215, ed. Kukol A. Ch. 3 (Humana Press: New York, NY, 2015).
Huang, J. & MacKerell, A. D. Force field development and simulations of intrinsically disordered proteins. Curr. Opin. Struct. Biol. 48, 40–48 (2018).
Dilabio, G. A. In Non-Covalent Interactions in Quantum Chemistry and Physics: Theory and Applications 1st edn, eds Otero-de-la-Roza A. & Dilabio G. A. Ch. 7 (Elsevier Inc., 2017).
Prasad, V. K., Otero-de-la-Roza, A. & DiLabio, G. A. Atom-centered potentials with dispersion-corrected minimal-basis-set Hartree–Fock: an efficient and accurate computational approach for large molecular systems. J. Chem. Theory Comput. 14, 726–738 (2018).
Kruse, H. & Grimme, S. A geometrical correction for the inter- and intra-molecular basis set superposition error in Hartree-Fock and density functional theory calculations for large systems. J. Chem. Phys. 136, 154101 (2012).
Řezáč, J. & Hobza, P. Advanced corrections of hydrogen bonding and dispersion for semiempirical quantum mechanical methods. J. Chem. Theory Comput. 8, 141–151 (2012).
Witte, J., Neaton, J. B. & Head-Gordon, M. Effective empirical corrections for basis set superposition error in the def2-SVPD basis: gCP and DFT-C. J. Chem. Phys. 146, 234105 (2017).
Lindorff-Larsen, K. et al. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct. Funct. Bioinforma 78, 1950–1958 (2010).
Maier, J. A. et al. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713 (2015).
Wang, L.-P. et al. Building a more predictive protein force field: a systematic and reproducible route to AMBER-FB15. J. Phys. Chem. B 121, 4023–4039 (2017).
MacKerell, A. D. Jr. et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616 (1998).
Best, R. B. et al. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ1 and χ2 dihedral angles. J. Chem. Theory Comput. 8, 3257–3273 (2012).
Huang, J. et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73 (2017).
Robertson, M. J., Tirado-Rives, J. & Jorgensen, W. L. Improved peptide and protein torsional energetics with the OPLS-AA force field. J. Chem. Theory Comput. 11, 3499–3509 (2015).
Shi, Y. et al. Polarizable atomic multipole-based AMOEBA force field for proteins. J. Chem. Theory Comput. 9, 4046–4063 (2013).
Thiel, W. Semiempirical quantum-chemical methods. Wiley Interdiscip. Rev. Comput. Mol. Sci 4, 145–157 (2014).
Brandenburg, J. G., Hochheim, M., Bredow, T. & Grimme, S. Low-cost quantum chemical methods for noncovalent interactions. J. Phys. Chem. Lett. 5, 4275–4284 (2014).
Christensen, A. S., Kubař, T., Cui, Q. & Elstner, M. Semiempirical quantum mechanical methods for noncovalent interactions for chemical and biochemical applications. Chem. Rev. 116, 5301–5337 (2016).
Dewar, M. J. S., Zoebisch, E. G., Healy, E. F. & Stewart, J. J. P. Development and use of quantum mechanical molecular models. 76. AM1: a new general purpose quantum mechanical molecular model. J. Am. Chem. Soc 107, 3902–3909 (1985).
Stewart, J. J. P. Optimization of parameters for semiempirical methods V: modification of NDDO approximations and application to 70 elements. J. Mol. Model. 13, 1173–1213 (2007).
Řezáč, J., Fanfrlík, J., Salahub, D. & Hobza, P. Semiempirical quantum chemical PM6 method augmented by dispersion and H-bonding correction terms reliably describes various types of noncovalent complexes. J. Chem. Theory Comput. 5, 1749–1760 (2009).
Stewart, J. J. P. Optimization of parameters for semiempirical methods VI: more modifications to the NDDO approximations and re-optimization of parameters. J. Mol. Model. 19, 1–32 (2013).
Tuttle, T. & Thiel, W. OMx-D: semiempirical methods with orthogonalization and dispersion corrections. Implementation and biochemical application. Phys. Chem. Chem. Phys. 10, 2159–2166 (2008).
Dral, P. O. et al. Semiempirical quantum-chemical orthogonalization-corrected methods: theory, implementation, and parameters. J. Chem. Theory Comput. 12, 1082–1096 (2016).
Frauenheim, T. H. et al. A self-consistent charge density-functional based tight-binding method for predictive materials simulations in physics, chemistry and biology. physica status solidi (b) 217, 41–62 (2000).
Koskinen, P. & Mäkinen, V. Density-functional tight-binding for beginners. Comput. Mater. Sci. 47, 237–253 (2009).
Krishnapriyan, A., Yang, P., Niklasson, A. M. N. & Cawkwell, M. J. Numerical optimization of density functional tight binding models: application to molecules containing carbon, hydrogen, nitrogen, and oxygen. J. Chem. Theory Comput. 13, 6191–6200 (2017).
Grimme, S., Bannwarth, C. & Shushkov, P. A robust and accurate tight-binding quantum chemical method for structures, vibrational frequencies, and noncovalent interactions of large molecular systems parametrized for all spd-block elements (Z = 1–86). J. Chem. Theory Comput. 13, 1989–2009 (2017).
Sure, R. & Grimme, S. Corrected small basis set Hartree-Fock method for large systems. J. Comput. Chem. 34, 1672–1685 (2013).
Goerigk, L. & Reimers, J. R. Efficient methods for the quantum chemical treatment of protein structures: the effects of London-dispersion and basis-set incompleteness on peptide and water-cluster geometries. J. Chem. Theory Comput. 9, 3240–3251 (2013).
Goerigk, L., Collyer, C. A. & Reimers, J. R. Recommending Hartree–Fock theory with London-dispersion and basis-set-superposition corrections for the optimization or quantum refinement of protein structures. J. Phys. Chem. B 118, 14612–14626 (2014).
Grimme, S., Brandenburg, J. G., Bannwarth, C. & Hansen, A. Consistent structures and interactions by density functional theory with small atomic orbital basis sets. J. Chem. Phys. 143, 054107 (2015).
Sure, R., Brandenburg, J. G. & Grimme, S. Small atomic orbital basis set first-principles quantum chemical methods for large molecular and periodic systems: a critical analysis of error sources. ChemistryOpen 5, 94–109 (2016).
Brandenburg, J. G., Caldeweyher, E. & Grimme, S. Screened exchange hybrid density functional for accurate and efficient structures and interaction energies. Phys. Chem. Chem. Phys. 18, 15519–15523 (2016).
Brandenburg, J. G., Bannwarth, C., Hansen, A. & Grimme, S. B97-3c: A revised low-cost variant of the B97-D density functional method. J. Chem. Phys. 148, 064104 (2018).
Hamley, I. W. Small bioactive peptides for biomaterials design and therapeutics. Chem. Rev. 117, 14015–14041 (2017).
Case, D. A. et al. AMBER. (University of California, 2016).
Case, D. A. et al. The Amber biomolecular simulation programs. J. Comput. Chem. 26, 1668–1688 (2005).
Salomon-Ferrer, R., Case, D. A. & Walker, R. C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci 3, 198–210 (2013).
RCSB Protein Data Bank. https://www.rcsb.org/ (2018).
Berman, H. M. et al. The protein data bank. Nucleic Acids Res 28, 235–242 (2000).
The Cambridge Structural Database. https://www.ccdc.cam.ac.uk/solutions/csd-system/components/csd/ (2018).
Groom, C. R., Bruno, I. J., Lightfoot, M. P. & Ward, S. C. The cambridge structural database. Acta Cryst. B 72, 171–179 (2016).
The Open Babel Package, version 2.3.2 http://openbabel.org (2018).
O’Boyle, N. M. et al. Open Babel: an open chemical toolbox. J. Cheminform. 3, 33 (2011).
Avogadro: An Advanced Molecule Editor and Visualizer. https://avogadro.cc/ (2018).
Hanwell, M. D. et al. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 4, 17 (2012).
Bruccoleri, R. E. & Karplus, M. Conformational sampling using high-temperature molecular dynamics. Biopolymers 29, 1847–1862 (1990).
Settanni, G. & Fersht, A. R. High temperature unfolding simulations of the TRPZ1 peptide. Biophys. J. 94, 4444–4453 (2008).
Walczewska-Szewc, K., Deplazes, E. & Corry, B. Comparing the ability of enhanced sampling molecular dynamics methods to reproduce the behavior of fluorescent labels on proteins. J. Chem. Theory Comput. 11, 3455–3465 (2015).
Dalby, A. & Shamsir, M. S. Molecular Dynamics Simulations of the Temperature Induced Unfolding of Crambin Follow the Arrhenius Equation. F1000Research 4, 589 (2015).
Neale, C., Pomès, R. & García, A. E. Peptide bond isomerization in high-temperature simulations. J. Chem. Theory Comput. 12, 1989–1999 (2016).
RDKit: Open-Source Cheminformatics Software, https://www.rdkit.org/ (2018).
Ebejer, J.-P., Morris, G. M. & Deane, C. M. Freely available conformer generation methods: how good are they? J. Chem. Inf. Model. 52, 1146–1158 (2012).
Blaney, J. M. & Dixon, J. S. In Reviews in Computational Chemistry Vol. 5, eds Lipkowitz, K. B. & Boyd, D. B. Ch. 6 (John Wiley & Sons, Inc., 2007).
Smith, J. S., Isayev, O. & Roitberg, A. E. ANI-1, A data set of 20 million calculated off-equilibrium conformations for organic molecules. Sci. Data 4, 170193 (2017).
Becke, A. D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 38, 3098–3100 (1988).
Lee, C., Yang, W. & Parr, R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785–789 (1988).
Hariharan, P. C. & Pople, J. A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 28, 213–222 (1973).
Francl, M. M. et al. Self‐consistent molecular orbital methods. XXIII. A polarization‐type basis set for second‐row elements. J. Chem. Phys 77, 3654–3665 (1982).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Grimme, S., Ehrlich, S. & Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32, 1456–1465 (2011).
Becke, A. D. & Johnson, E. R. Exchange-hole dipole moment and the dispersion interaction. J. Chem. Phys. 122, 154104 (2005).
Johnson, E. R. & Becke, A. D. A post-Hartree–Fock model of intermolecular interactions. J. Chem. Phys. 123, 024101 (2005).
Becke, A. D. & Johnson, E. R. A density-functional model of the dispersion interaction. J. Chem. Phys. 123, 154101 (2005).
Becke, A. D. & Johnson, E. R. Exchange-hole dipole moment and the dispersion interaction: high-order dispersion coefficients. J. Chem. Phys. 124, 014104 (2006).
Johnson, E. R. & Becke, A. D. A post-Hartree-Fock model of intermolecular interactions: inclusion of higher-order corrections. J. Chem. Phys. 124, 174104 (2006).
Becke, A. D. & Johnson, E. R. A unified density-functional treatment of dynamical, nondynamical, and dispersion correlations. J. Chem. Phys. 127, 124108 (2007).
Becke, A. D. & Johnson, E. R. Exchange-hole dipole moment and the dispersion interaction revisited. J. Chem. Phys. 127, 154108 (2007).
Otero-de-la-Roza, A. & DiLabio, G. A. Transferable atom-centered potentials for the correction of basis set incompleteness errors in density-functional theory. J. Chem. Theory Comput. 13, 3505–3524 (2017).
Frisch, M. J. et al. Gaussian 09, Revision D.01. (Gaussian, Inc., Wallingford CT, 2009).
Frisch, M. J. et al. Gaussian 16, Revision B.01. (Gaussian, Inc., Wallingford CT, 2016).
Huzinaga, S. Basis sets for molecular calculations. Comput. Phys. Rep. 2, 281–339 (1985).
Vydrov, O. A. & Scuseria, G. E. Assessment of a long-range corrected hybrid functional. J. Chem. Phys. 125, 234109 (2006).
Vydrov, O. A., Heyd, J., Krukau, A. V. & Scuseria, G. E. Importance of short-range versus long-range Hartree-Fock exchange for the performance of hybrid density functionals. J. Chem. Phys. 125, 074106 (2006).
Dunning, T. H. Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 90, 1007–1023 (1989).
Kendall, R. A., Dunning, T. H. Jr. & Harrison, R. J. Electron affinities of the first‐row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys 96, 6796–6806 (1992).
Woon, D. E. & Dunning, T. H. Jr. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 98, 1358–1371 (1993).
Otero-de-la-Roza, A., Johnson, E. R. & DiLabio, G. A. Halogen bonding from dispersion-corrected density-functional theory: the role of delocalization error. J. Chem. Theory Comput. 10, 5436–5447 (2014).
Hohenstein, E. G. & Sherrill, C. D. Wavefunction methods for noncovalent interactions. Wiley Interdiscip. Rev. Comput. Mol. Sci 2, 304–326 (2012).
Řezáč, J. & Hobza, P. Describing noncovalent interactions beyond the common approximations: how accurate is the “Gold Standard”, CCSD(T) at the complete basis set limit? J. Chem. Theory Comput. 9, 2151–2155 (2013).
Riplinger, C. & Neese, F. An efficient and near linear scaling pair natural orbital based local coupled cluster method. J. Chem. Phys. 138, 034106 (2013).
Riplinger, C., Sandhoefer, B., Hansen, A. & Neese, F. Natural triple excitations in local coupled cluster calculations with pair natural orbitals. J. Chem. Phys. 139, 134101 (2013).
Gruzman, D., Karton, A. & Martin, J. M. L. Performance of ab initio and density functional methods for conformational equilibria of C nH2n+2 alkane isomers (n = 4−8). J. Phys. Chem. A 113, 11974–11983 (2009).
Kozuch, S., Bachrach, S. M. & Martin, J. M. L. Conformational equilibria in butane-1,4-diol: a benchmark of a prototypical system with strong intramolecular H-bonds. J. Phys. Chem. A 118, 293–303 (2014).
Marianski, M., Supady, A., Ingram, T., Schneider, M. & Baldauf, C. Assessing the accuracy of across-the-scale methods for predicting carbohydrate conformational energies for the examples of glucose and α-maltose. J. Chem. Theory Comput. 12, 6157–6168 (2016).
Fogueri, U. R., Kozuch, S., Karton, A. & Martin, J. M. L. The melatonin conformer space: benchmark and assessment of wave function and DFT methods for a paradigmatic biological and pharmacological molecule. J. Phys. Chem. A 117, 2269–2277 (2013).
Csonka, G. I., French, A. D., Johnson, G. P. & Stortz, C. A. Evaluation of density functionals and basis sets for carbohydrates. J. Chem. Theory Comput. 5, 679–692 (2009).
Kruse, H. et al. Quantum chemical benchmark study on 46 RNA backbone families using a dinucleotide unit. J. Chem. Theory Comput. 11, 4972–4991 (2015).
Burns, L. A. et al. The biofragment database (BFDb): an open-data platform for computational chemistry analysis of noncovalent interactions. J. Chem. Phys. 147, 161727 (2017).
Jurečka, P., Šponer, J., Černý, J. & Hobza, P. Benchmark database of accurate (MP2 and CCSD(T) complete basis set limit) interaction energies of small model complexes, DNA base pairs, and amino acid pairs. Phys. Chem. Chem. Phys. 8, 1985–1993 (2006).
Gráfová, L., Pitoňák, M., Řezáč, J. & Hobza, P. Comparative study of selected wave function and density functional methods for noncovalent interaction energy calculations using the extended S22 data set. J. Chem. Theory Comput. 6, 2365–2376 (2010).
Řezáč, J., Riley, K. E. & Hobza, P. S66: A well-balanced database of benchmark interaction energies relevant to biomolecular structures. J. Chem. Theory Comput. 7, 2427–2438 (2011).
Řezáč, J., Riley, K. E. & Hobza, P. Extensions of the S66 data set: more accurate interaction energies and angular-displaced nonequilibrium geometries. J. Chem. Theory Comput. 7, 3466–3470 (2011).
Brauer, B., Kesharwani, M. K., Kozuch, S. & Martin, J. M. L. The S66x8 benchmark for noncovalent interactions revisited: explicitly correlated ab initio methods and density functional theory. Phys. Chem. Chem. Phys. 18, 20905–20925 (2016).
Otero-de-la-Roza, A. & Johnson, E. R. Non-covalent interactions and thermochemistry using XDM-corrected hybrid and range-separated hybrid density functionals. J. Chem. Phys. 138, 204109 (2013).
Data Citations
Prasad, V. K., Otero-de-la-Roza, A., & DiLabio, G. A. figshare https://doi.org/10.6084/m9.figshare.7185194.v2 (2018)
Acknowledgements
GAD would like to thank the Natural Sciences and Engineering Research Council of Canada, the Canadian Foundation for Innovation, and the British Columbia Knowledge Development Fund for financial support. We are grateful to Compute Canada/Westgrid for a generous allocation of computing resources.
Author information
Authors and Affiliations
Contributions
V.K.P. performed the selection, calculation, curation and validation associated with the generation of the PEPCONF data set. A.O.R. assisted in the calculation, curation, and post-validation checks. G.A.D. designed the idea of the study and cross-validated the final results. V.K.P., A.O.R., and G.A.D. co-wrote the data descriptor.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Supplementary information accompanies this paper at
ISA-Tab metadata
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files made available in this article.
About this article

Cite this article
Prasad, V., Otero-de-la-Roza, A. & DiLabio, G. PEPCONF, a diverse data set of peptide conformational energies. Sci Data 6, 180310 (2019). https://doi.org/10.1038/sdata.2018.310
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/sdata.2018.310
This article is cited by
-
Estimating the Influence of Physicochemical and Biochemical Property Indexes on Selection for Amino Acids Usage in Eukaryotic Cells
Journal of Molecular Evolution (2021)
