
Abstract
Leptosphaeria maculans and Leptosphaeria biglobosa are ascomycete phytopathogens of Brassica napus (oilseed rape, canola). Here we report the complete sequence of three Leptosphaeria genomes (L. maculans JN3, L. maculans Nz-T4 and L. biglobosa G12-14). Nz-T4 and G12-14 genome assemblies were generated de novo and the reference JN3 genome assembly was improved using Oxford Nanopore MinION reads. The new assembly of L. biglobosa showed the existence of AT rich regions and pointed to a genome compartmentalization previously unsuspected following Illumina sequencing. Moreover nanopore sequencing allowed us to generate a chromosome-level assembly for the L. maculans reference isolate, JN3. The genome annotation was supported by integrating conserved proteins and RNA sequencing from Leptosphaeria-infected samples. The newly produced high-quality assemblies and annotations of those three Leptosphaeria genomes will allow further studies, notably focused on the tripartite interaction between L. maculans, L. biglobosa and oilseed rape. The discovery of as yet unknown effectors will notably allow progress in B. napus breeding towards L. maculans resistance.
Design Type(s) | sequence assembly objective • transcription profiling by high throughput sequencing design • sequence annotation objective |
Measurement Type(s) | genome assembly • sequence annotation |
Technology Type(s) | whole genome sequencing • RNA sequencing |
Factor Type(s) | Organism Strain • Biospecimen Type • cultivar • culture medium • sampling time |
Sample Characteristic(s) | Leptosphaeria maculans 'brassicae' group • Leptosphaeria biglobosa 'brassicae' group • Leptosphaeria maculans species complex |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others
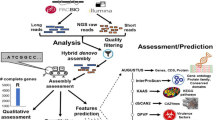

Background & Summary
Fungi have an important ecological and economic role. Many of them are pathogens of animals or plants, especially Leptosphaeria maculans which is an ascomycete phytopathogen of Brassica napus (oilseed rape, canola). Two genomes of Leptosphaeria were already available, the first genome sequence of this fungus (JN3) was obtained in the 2000’s using Sanger sequencing1 and composed of 41 scaffolds totaling 43.76 Mb (excluding ambiguous bases). In-depth analysis of this genome showed an unusual compartmentalization between AT-rich and GC-equilibrated blocks termed isochores. The location of effector genes in the plastic, AT-rich compartments of the genome was instrumental in establishing the “two-speed fungal genome” paradigm1. According to this paradigm, phytopathogen genomes encompass a plastic compartment enriched in genes involved in niche adaptation. Similar genome organisation was later on found to be a commonality in numerous filamentous phytopathogens, but its identification was slowed down by the use of short-reads sequencing that tended to misrepresent the plastic, Transposable Element (TE)-rich genome in the assemblies. Thus the genome sequencing of Leptosphaeria biglobosa (B3.5), a species related to L. maculans resulted in a poor-quality assembly with complete absence of a putative dispensable genome2. This assembly of B3.5 was composed of 606 scaffolds totaling 29.44 Mb (excluding ambiguous bases).
Generally, when using short-read sequencing strategies, TE complicate the assembly step and it results in an under estimation of the repetitive content by collapsing similar copies of repetitions. In this study, we generated genome assembly and annotation of three Leptosphaeria isolates. To overcome the limitation of short reads, we generated long reads using the Oxford Nanopore technology (Data Citation 1, 2, 3). First, we resequenced the JN3 isolate with the objective of improving the existing assembly in terms of both contiguity and gaps closure (Fig. 1). And then we de novo sequenced and assembled two isolates L. maculans Nz-T4 and L. biglobosa G12-14 (Fig. 1). For each isolate, we generated long and short reads using respectively the MinION device from Oxford Nanopore and the Illumina sequencing technology (Tables 1, 2, 3). Reconciling manually the data from nanopore sequencing, optical and genetic maps allowed the generation of a chromosome-scale assembly for JN3 composed of 19 chromosomes plus the conditionally dispensable chromosome3, with only four missing telomeric extremities. The three genome assemblies were composed of 33, 288 and 156 scaffolds totaling 45.99 Mb, 43.42 Mb and 34.95 Mb for JN3, Nz-T4 and G12-14, respectively (Table 4). Furthermore, the gene prediction was supported by integrating conserved proteins and RNA sequencing from Leptosphaeria-infected samples (Fig. 2 and Data Citation 4). It resulted in 13,047, 14,026 and 12,678 predicted protein-coding genes for JN3, Nz-T4 and G12-14, respectively (Table 5 and Fig. 3).

Amelioration of the existing JN3 assembly and de novo assembly of Nz-T4 and G12-14 isolates.

General description of the gene prediction workflow.

The genome browser is available at http://www.genoscope.cns.fr/leptolife and contains repeats (green track), coverage of genomic reads (black wiggle), gene prediction (blue track), RNA contigs (dark green track) and protein homologies (salmon track).
Methods
Biological material
Two L. maculans brassicae isolates, JN3 (v23.1.3) and Nz-T4 were sequenced here. JN3 is the reference sequenced isolate1 of European origin. Nz-T4 has been used in previous genetic analyses4 and originates from New Zealand. The L. biglobosa brassicae isolate G12-14, also sequenced here, was isolated in 2014 in France (Thiverval-Grignon) as a single-ascospore isolate from naturally infected oilseed rape stubble. G12-14 was chosen as a new L. biglobosa reference isolate since the previously sequenced isolate, B3.52, showed a very low level of aggressiveness when used in cotyledon inoculation tests. In contrast, G12-14 rapidly produced on oilseed rape plants typical and intense symptoms of L. biglobosa brassicae. In addition it was considered as a better representative of the current L. biglobosa populations infecting oilseed rape in France than B3.5.
For RNA sequencing, additional isolates were used, including L. maculans JN2 (v23.1.2), a sister isolate of JN3 with a high level of aggressiveness and the L. biglobosa brassicae reference isolate, B3.52. Plant materials infected with L. maculans and/or L. biglobosa were obtained under various conditions (in the field or under controlled conditions) and with three different B. napus varieties. Naturally infected plant material was obtained in Thiverval-Grignon experimental fields as either infected stem base at the end of the growing season, sampled one week before harvest (variety Darmor-Bzh) or as whole stem residues sampled two weeks after harvest (variety Alpaga). Under controlled conditions, two varieties were used, Darmor-Bzh displaying a high level of field resistance to L. maculans, and Bristol, with a high level of susceptibility. Cotyledons and stems were inoculated as previously described5 and inoculated material was sampled at different time points after inoculation. For petiole inoculations, the petiole was cut under the leaf blade. Ten μL of inoculum (107 pycnidiospores mL−1) were placed over the wound. Inoculated plants were incubated in darkness for 48 h at room temperature under saturated humidity, and then transferred to a growth chamber at 16 °C (night) and 24 °C (day), with a 16 h photoperiod. Infected petioles were sampled after 7 and 14 days post infection (dpi).
RNA/DNA extraction
Fresh infected cotyledons or freeze-dried cultures of fungal mycelia were ground using a Retsch MM300 mixer mill in Eppendorf tubes, with one tungsten carbide bead per tube, for 45 s with 30 oscillations per second. Petioles were ground with a pestle and mortar. Stem base and stem residues were ground with a Retsch MM300 mixer mill using Zirconium oxide jars and beads, for 40 s with 30 oscillations per second. For RNA extraction, all grinding material was frozen with liquid nitrogen prior grinding. RNA extraction was then performed using the Trizol reagent (Invitrogen, Cergy Pontoise, France) as previously described6. DNA was extracted from freeze-dried material using the plant DNeasy mini-kit or the Wizard® Genomic DNA Purification Kit (Promega, Madison, WI, USA) according to the manufacturers’ instructions. Input DNA for the nanopore sequencing, based on long DNA molecules, was extracted from agarose plugs of concentrated pycnidiospores (>1.109 spore ml−1) partially digested1,3. Briefly, fungal pycnidiospores were collected from V8-agar plates3 with sterile distilled water, centrifuged (15 min at 10000×g) and adjusted to 6.109 spore mL1. The spore suspension was mixed with an equal volume of 2.5% low melting agarose (Seaplaque GTG® in TSE; Tris 25 mM, pH 7.5; Sorbitol 1 M, EDTA 25 mM) maintained at 40 °C. This suspension was poured in disposable plug molds (BioRad). After cooling, the resulting plugs were treated for 20 h in 0.5 M EDTA, SDS 10%, 1 mg.mL−1 proteinase K (Sigma) at 50 °C under mild stirring, then rinsed three times with 0.5 M EDTA, 50 °C and stored at 4 °C in 0.5 M EDTA. For a part of Nanopore sequencing runs, DNA was also extracted from freeze-dried mycelium ground in liquid nitrogen: cells have been then lysed using CTAB (Cetyltrimethylammonium bromide) with 2% of PVP (Polyvinylpyrrolidone) and 0.1% of 2-mercaptoéthanol and DNA was purified following a phenol-chloroform protocol.
Illumina PCR-Free library preparation and sequencing
DNA (4.5 to 6 μg) was sonicated to a 100- to 1500-bp size range using a Covaris E210 sonicator (Covaris, Woburn, MA, USA). Fragments were end-repaired using the NEBNext End Repair Module (New England Biolabs, Ipswich, MA, USA) and 3΄-adenylated with the NEBNext dA-Tailing Module (New England Biolabs). Illumina adapters were then added using the NEBNext Quick Ligation Module (New England Biolabs) and ligation products were purified with AMPure XP beads (Beckmann Coulter Genomics, Danvers, MA, USA). Libraries were sequenced on an Illumina MiSeq (G12-14 and NzT4 genomes) or a HiSeq 2500 (JN3 genome) instrument (San Diego, CA, USA) using 250 base-length read chemistry in a paired-end mode.
Illumina cDNA library preparation and sequencing
RNA-Seq library preparations were carried out from 1–2 μg total RNA using the TruSeq Stranded mRNA kit (Illumina, San Diego, CA, USA), which allows mRNA strand orientation (sequence reads occur in the same orientation as anti-sense RNA). Briefly, poly(A)+ RNA was selected with oligo(dT) beads, chemically fragmented and converted into single-stranded cDNA using random hexamer priming. Then, the second strand was generated to create double-stranded cDNA. cDNA were then 3′-adenylated, and Illumina adapters were added. Ligation products were PCR-amplified. Each library was sequenced using 101 bp paired end reads chemistry on a HiSeq2000 Illumina sequencer.
Nanopore 8-kb and 20-kb libraries preparation
MinION sequencing libraries were prepared according to the nanopore Sequencing Kit protocol SQK-MAP006 or the Low Input Expansion Pack protocol for genomic DNA. Briefly, 100 ng to 1.5 μg of genomic DNA was sheared to approximately 8 Kb with g-TUBE (Covaris, Woburn, MA, USA) and cleaned-up using AMPure XP beads (Beckmann Coulter Genomics). For some libraries, DNA fragments were repaired using the NEBNext FFPE Repair Mix (New England Biolabs). After end-reparation and 3′-adenylation with the NEBNext® UltraTM II End Repair/dA-Tailing Module (New England Biolabs), sequencing adapters provided by Oxford Nanopore Technologies (Oxford Nanopore Technologies Ltd, UK) were ligated using Blunt/TA Ligase Master Mix (New England Biolabs). Libraries were then enriched using Dynabeads MyOne C1 Streptavidin (ThermoFisher Scientific, Wilmington, DE, USA).
Nanopore 20-Kb libraries were prepared according to the same protocol, with the exception that 250 ng to 2.5 μg of genomic DNA was sheared to approximately 20-kb with g-TUBE (Covaris).
MinION flow cell preparation and sample loading
Each library was mixed with the fuel mix and the running buffer according to the SQK-MAP006 or the Low Input Expansion Pack protocols. The sequencing mix was then added to the R7.3 flowcell for a 48-hour run. Several loading schedules were tested, but the main one used for the MAP006 libraries has been the following: a first loading with 8 μl of the library, then three reloading after 5, 24 and 29 h of run with respectively 4 μl, 8 μl and 4 μl of the DNA library. Regarding the low-input libraries, the main schedule used was a first loading with 10 μl of the DNA library and a reloading after 24 hours of run time with 10 μl of the DNA library.
MinION sequencing and basecalling
Read event data generated by MinKNOW control software (version 0.50.2.15 to 0.51.3.40) were base-called using the Metrichor software (version 2.34.3 to 2.39.3). The data generated (pores metrics, sequencing, and base-calling data) by MinION software were stored and organized using a Hierarchical Data Format. Three types of reads were obtained: template, complement, and two-directions (2D). The template and complement reads correspond to sequencing of the two DNA strands. Metrichor combines template and complement reads to produce a consensus (2D) sequence7. FASTA reads were extracted from MinION Hierarchical Data Format files using poretools8. Pass and fail reads were both considered as 1D reads. This dataset is described in Table 1 and (Genomic datasets, Data Citation 5).
Genetic map
A genetic map was built using 93 progeny of the JN2×Nz-T4 in vitro cross9. A total of 150 polymorphic micro- and minisatellite markers, three biological markers (Mat locus, AvrLm3 and AvrLm7) and eight markers designed at the junction between two repeated elements10 were first used to generate an unsaturated genetic map using Mapmaker 3.0, with a LOD score of 3 and a maximum genetic distance of 20 cM. The 93 progeny isolates and the two parental isolates were also sequenced (2 × 100 bp illumina sequencing; 48 samples per line; Beckman Coulter Genomics). SNP calling between parental isolates was done as described in the study11 following mapping of the reads on the JN3 reference genome. A total of 20,103 reliable SNPs were detected and assigned to one of the two parental isolate in the progeny. These SNPs were converted into 2,104 Bins. From all these markers, a saturated map comprising 20 linkage groups was constructed using MSTmap online.
Long reads-based de novo assembly of L. biglobosa G12-14 and L. maculans Nz-T4
R7.3 nanopore 2D reads (Table 3) were assembled into contigs using SmartDeNovo (https://github.com/ruanjue/smartdenovo), with a k-mer size parameter set to 21. Eventually, the consensus of the assembly was polished by aligning the Illumina paired-end 2 × 250 bp reads (Table 2) with BWA mem12 and using the resulting alignment file as input for Pilon13. We iteratively polished twice the assembly.
L. biglobosa G12-14 final assembly had a cumulative size of 34.9 Mb and a N50 equal to 462 kb. L. maculans Nz-T4 final assembly had a cumulative size of 43.4 Mb and a N50 equal to 383 kb. Further usual metrics regarding those assemblies are provided in Table 4.
Genome assembly update of L. maculans JN3
With 33 scaffolds, a N50 equal to 2.40 Mb and a cumulative size of 44.8 Mb, the JN3 rearranged reference genome was already contiguous enough for our analyses. However, it contained gaps that represented 1.1 Mb in total. In addition, both the optical map2 and the high-density genetic map indicated that nine mis-assembled super contigs had to be either splitted or fused. The reference genome was scaffolded and gap-filled using PBJelly14 with the entire nanopore dataset as input (Table 3). As a result we obtained 33 scaffolds with a cumulative size of 45.8 Mb, the N50 increased to 2.43 Mb and we decreased by half the number of N’s (570 kb).
As the R7.3 nanopore reads were prone to errors, particularly in the homopolymeric regions15, we polished the consensus of the gap closed assembly by aligning 150X of Illumina paired-end 2 × 250 bp reads with BWA mem12 and giving the resulting alignment file to Pilon13. The final assembly had a cumulative size of 45.99 Mb with 1% of unknown bases (Table 4).
Genome annotation
The gene prediction of the three Leptosphaeria genomes was done by combining gene models information computed from transcript and protein alignments (Fig. 2).
First, repeated sequences in the reference genomes were masked using multiple tools. Tandem repeats were identified and masked using Tandem Repeats Finder16. Low complexity regions were identified and masked using Dust17. Interspersed repeats and other low complexity sequences were masked using RepeatMasker (http://www.repeatmasker.org). Furthermore 121 known transposable elements (identified in the L. maculans-L. biglobosa species complex in a previous study2) were also masked using RepeatMasker.
The Alternaria alternata, Cochliobolus heterostrophus, Pyrenochaeta sp., Pyrenophora tritici-repentis and Zymoseptoria tritici proteomes included in UniProt (version available as of 07 September 2016) were used to detect conserved proteins in L. maculans JN3, L. maculans Nz-T4 and L. biglobosa G12-14. L. maculans JN3 and swissprot proteins (release available as of 30 November 2016) were also used as a resource to detect conserved proteins in L. maculans Nz-T4 and L. biglobosa G12-14.
Those proteins were first aligned to L. maculans JN3, L. maculans Nz-T4 and L. biglobosa G12-14 genomes assemblies using BLAT18. BLAT results were then filtered as follows: for each protein, the best match (based on the BLAT score) and other matches with a score > = 90% of the score of the best match were retained. Genewise19 was then used to refine matches and identify exon-intron boundaries and alignment were filtered out if less than 50% of the protein length was aligned.
RNAseq reads from 23 samples were assembled using Velvet20 1.2.07 and Oases21 0.2.08, using a k-mer size of 63 bp. The results of this assembly for each sample are summarized in (Transcriptomic datasets, Data Citation 5). Reads were mapped back to the contigs with BWA-mem12 and the consistent paired-end reads were selected. Chimeric contigs were identified and splitted (uncovered regions) based on coverage information from consistent paired-end reads. Moreover, open reading frames (ORF) and domains were searched using respectively TransDecoder22 and CDDsearch23. We only allowed breaks outside ORF and domains. At the end, the read strand information was used to correctly orient the RNA-Seq contigs. In a second step, RNAseq contigs were aligned to the relevant genome assembly (L. maculans JN3, L. maculans Nz-T4, L. biglobosa G12-14) using BLAT. The best matches (based on BLAT score) for each contig and other matches with a score > = 90% of the score of the best match were selected. Then, Est2genome24 was used to refine the alignments and we kept alignments with an identity percent > = 95% and at least 80% of the RNA-Seq contig that can be aligned (Transcriptomic datasets, Data Citation 5).
Gene predictions from mRNA and proteomes were given as input to the GMOVE combiner25 to build the gene models. This tool considers biological data such as RNA-seq from the organisms of interest and proteome from homologous species. Proteins give information about the frame of the coding sequence (CDS) and RNA-seq helps to find splicing sites with more accurate alignments. This tool is designed for all eukaryotes and de novo studies with no need of calibration pre-processing. After gene models were produced by GMOVE, a final filter was applied to remove artefactual untranslated regions (UTRs). UTRs were trimmed if it overlapped CDS in predicted gene models for more than 10 bases. Moreover, UTRs overlapping other UTRs for more than 10 bases were also trimmed to avoid large and potentially false UTRs. Final metrics of the gene prediction are available in Table 5 and Fig. 3 is a screenshot of the genome browser.
Code availability
All the tools used for this study are cited in the method section and they were used with default settings unless options were specified. The in-house tools to clean illumina data are available from the following website: http://www.genoscope.cns.fr/fastxtend/. The integration of resources in the gene prediction workflow was achieved using GMOVE: http://www.genoscope.cns.fr/gmove/.
Data Records
The authors declare that all data reported herein are fully and freely available from the date of publication. Genomic data have been deposited in the ENA repository (Genomic datasets, Data Citation 5): L. maculans JN3 (Data Citation 1), L. maculans Nz-T4 (Data Citation 2) and L. biglobosa G12-14 (Data Citation 3). RNA data used for gene prediction have been deposited in the ENA repository (Data Citation 4 and Transcriptomic datasets, Data Citation 5). Moreover, a dedicated website brings together all the information: accession numbers, assembly and annotation files as well as a genome browser (Fig. 3): http://www.genoscope.cns.fr/leptolife.
Technical Validation
DNA and RNA sample quality
DNA quality was assessed using 1% agarose gel. RNA integrity was assessed using an Agilent 2100 Bioanalyzer electrophoresis system (Agilent, Santa Clara, CA, USA).
Illumina libraries
Ready-to-sequence Illumina libraries were quantified by qPCR using the KAPA Library Quantification Kit for Illumina Libraries (KapaBiosystems, Wilmington, MA, USA), and libraries profiles evaluated with an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA).
Illumina reads processing and quality filtering
After the Illumina sequencing, an in-house quality control process was applied to the reads that passed the Illumina quality filters. The first step discards low-quality nucleotides (Q < 20) from both ends of the reads. Next, Illumina sequencing adapters and primer sequences were removed from the reads. Then, reads shorter than 30 nucleotides after trimming were discarded. These trimming and removal steps were achieved using in-house-designed software based on the FastX package. The last step identifies and discards read pairs that mapped to the phage phiX genome, using SOAP26 and the phiX reference sequence (GenBank: NC_001422.1). This processing resulted in high-quality data and improvement of the subsequent analyses. This dataset is described in Table 2 and (Genomic datasets, Data Citation 5). A specific filter aiming to remove ribosomal reads was applied to data generated from RNA samples. The reads and their mates that mapped onto a ribosomal sequences database were filtered using SortMeRNA27. It contains different rRNA databases and we used it to split the data into two files: rRNA reads in a file (ribo_clean) and other reads in another file (noribo_clean). This dataset is described in (Transcriptomic datasets, Data Citation 5).
Impact of the gap closing procedure on the JN3 assembly
The JN3 assembly was gap closed using PBJelly and we detected several duplicated regions of several Kb in this gap-closed assembly that were not present in the previous assembly. We first detected tandemly duplicated genes using Blast and revealed 28 candidate regions totaling 150 kb in length with a median size of 5.5 kb and covering 63 genes. Further investigations of these regions using genomic data coverage revealed that they were artefactual duplicates created by PBJelly. We mapped both Illumina and nanopore reads onto the gap-closed assembly and highlighted genomic regions without illumina and nanopore reads coverage. This method revealed 30 regions with a cumulative size of 185 kb and a median size of 5.1 kb spanning a total of 43 genes. Finally, after a careful expertise using the genome browser (Fig. 3), the genomic regions that presented duplicated genes and no proper read support were masked and overlapping genes were filtered out. In total, we masked 41 regions with a cumulative size of 264 kb and a median size of 5.5 kb that spanned 81 genes (Duplicated genomic regions, Data Citation 5).
Contamination detection
In order to detect possible contamination in our assemblies, we first ran the metagene28 gene predictor on NzT4 and G12-14 contigs and on JN3 scaffolds. Then, we aligned all predicted proteins against the NR database using blastp. Hits were then filtered by their e-value to only conserve the best hit with an e-value of at least 10−10. Finally, a contig or scaffold was flagged as non-contaminant if at least 50% of its predicted proteins were assigned as fungi. Following this procedure, we couldn’t detect any contaminant sequences in JN3 and G12-14 assemblies. We detected 13 small contigs that were not assigned as fungi in the NzT4 assembly, but further investigations (blat against the JN3 assembly and blastx using the NCBI blast server) revealed that they were not contaminants.
Comparison with existing assemblies
An AT/GC region segmentation was performed on all assemblies with OcculterCut29 to detect potential genome compartmentalization as described in the following study1. L. maculans JN3 and Nz-T4 isolates showed similar genome architecture with a third of their assembly dispatched into AT blocks of 30.1 kb and 18.6 kb on average, respectively. The number of genes present within the AT-blocks was also steady, and ranged between 233 and 236. In contrast, there was a drastic modification in genome architecture and genome size between L. biglobosa B3.5, sequenced using only Illumina, and G12-14 sequenced here. The two assemblies differed by more than 10% of their genome content. This difference corresponded to 3 Mb missing in B3.5 and recovered in G12-14 thanks to the nanopore long reads (Table 6). The difference mostly comprised of AT-blocks similar to those found in L. maculans and amounting to 15.5% of the G12-14 assembly vs. only 4.9% in the B3.5 assembly (Table 6). The increase in AT-block content is consistent with the lowering of the average GC content of the L. biglobosa genome in the new assembly (Table 4).
Assessment of gene prediction
First, protein sequences of each genome (L. maculans JN3, L. maculans Nz-T4 and L. biglobosa G12-14) were obtained by translating gene CDS and aligned to protein databases (uniprot_sprot and uniprot_trembl 2016_11 release however L. maculans matches were excluded to prevent the introduction of known biases from the previous annotation effort) using blastp. Gene models for which proteins aligned with an e-value < 10−10 or with a CDS size > = 200 nt were retained.
Moreover gene completion for the newly generated annotations was evaluated using BUSCO30 v1.1b1 fungal and eukaryotic conserved genes databases. The relevance in the increase in gene number between the “old” and “new” gene prediction reflects a higher accuracy of the “new” annotation as evidenced by careful examination of genes encoding effectors. Effector genes encode for small (less than 300 amino-acids) secreted proteins (SSPs). They are usually poorly predicted de novo and may lack transcriptomic support due to expression in certain stages of parasitism only. The data obtained here drastically increased the number of predicted SSPs in both species (1,082 for L. maculans vs. 651 in the “old” annotation, 875 for L. biglobosa vs. 665 in the previous annotation) and drastically improved the gene models (data not shown). Also this new annotation allowed the identification of 75 genes hosted within the AT-block of the genome of L. biglobosa, previously unidentified (Table 6).
Additional information
How to cite this article: Dutreux, F. et al. De novo assembly and annotation of three Leptosphaeria genomes using Oxford Nanopore MinION sequencing. Sci. Data. 5:180235 doi: 10.1038/sdata.2018.235 (2018).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
References
Rouxel, T. et al. Effector diversification within compartments of the Leptosphaeria maculans genome affected by Repeat-Induced Point mutations. Nature communications 2, 202, 10.1038/ncomms1189 (2011).
Grandaubert, J. et al. Transposable element-assisted evolution and adaptation to host plant within the Leptosphaeria maculans Leptosphaeria biglobosa species complex of fungal pathogens. BMC genomics 15, 891, 10.1186/1471-2164-15-891 (2014).
Leclair, S., Ansan-Melayah, D., Rouxel, T. & Balesdent, M. Meiotic behaviour of the minichromosome in the phytopathogenic ascomycete Leptosphaeria maculans. Current genetics 30, 541–548 (1996).
Parlange, F. et al. Leptosphaeria maculans avirulence gene AvrLm4-7 confers a dual recognition specificity by the Rlm4 and Rlm7 resistance genes of oilseed rape, and circumvents Rlm4-mediated recognition through a single amino acid change. Molecular microbiology 71, 851–863, 10.1111/j.1365-2958.2008.06547.x (2009).
Gervais, J. et al. Different waves of effector genes with contrasted genomic location are expressed by Leptosphaeria maculans during cotyledon and stem colonization of oilseed rape. Molecular plant pathology 18, 1113–1126, 10.1111/mpp.12464 (2017).
Fudal, I. et al. Heterochromatin-like regions as ecological niches for avirulence genes in the Leptosphaeria maculans genome: map-based cloning of AvrLm6. Mol plant-microbe interact.: MPMI 20, 459–470, 10.1094/MPMI-20-4-0459 (2007).
Quick, J., Quinlan, A. R. & Loman, N. J. A reference bacterial genome dataset generated on the MinION portable single-molecule nanopore sequencer. GigaScience 3, 22, 10.1186/2047-217X-3-22 (2014).
Loman, N. J. & Quinlan, A. R. Poretools: a toolkit for analyzing nanopore sequence data. Bioinformatics 30, 3399–3401, 10.1093/bioinformatics/btu555 (2014).
Kuhn, M. L. et al. Genetic linkage maps and genomic organization in Leptosphaeria maculans. Eur J Plant Pathol 114, 17–31, 10.1007/s10658-005-3168-6 (2006).
Plissonneau, C. et al. A game of hide and seek between avirulence genes AvrLm4-7 and AvrLm3 in Leptosphaeria maculans. New Phytologist 209, 1613–1624, 10.1111/nph.13736 (2016).
Zhong, Z. et al. A small secreted protein in Zymoseptoria tritici is responsible for avirulence on wheat cultivars carrying the Stb6 resistance gene. New Phytologist 214, 619–631, 10.1111/nph.14434 (2017).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760, 10.1093/bioinformatics/btp324 (2009).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS one 9, e112963, 10.1371/journal.pone.0112963 (2014).
English, A. C. et al. Mind the gap: upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PloS one 7, e47768, 10.1371/journal.pone.0047768 (2012).
Istace, B. et al. de novo assembly and population genomic survey of natural yeast isolates with the Oxford Nanopore MinION sequencer. GigaScience 6, 1–13, 10.1093/gigascience/giw018 (2017).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic acids research 27, 573–580 (1999).
Morgulis, A., Gertz, E. M., Schaffer, A. A. & Agarwala, R. A fast and symmetric DUST implementation to mask low-complexity DNA sequences. Journal of Computational Biology 13, 1028–1040, 10.1089/cmb.2006.13.1028 (2006).
Kent, W. J. BLAT--the BLAST-like alignment tool. Genome Res. 12, 656–664, 10.1101/gr.229202 (2002).
Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome Res. 14, 988–995, 10.1101/gr.1865504 (2004).
Zerbino, D. R. & Birney, E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829, 10.1101/gr.074492.107 (2008).
Schulz, M. H., Zerbino, D. R., Vingron, M. & Birney, E. Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 28, 1086–1092, 10.1093/bioinformatics/bts094 (2012).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nature protocols 8, 1494–1512, 10.1038/nprot.2013.084 (2013).
Marchler-Bauer, A. & Bryant, S. H. CD-Search: protein domain annotations on the fly. Nucleic acids research 32, W327–W331, 10.1093/nar/gkh454 (2004).
Mott, R. EST_GENOME: a program to align spliced DNA sequences to unspliced genomic DNA. Computer applications in the biosciences: CABIOS 13, 477–478 (1997).
Dubarry, M, N. B. et al. Gmove a tool for eukaryotic gene predictions using various evidences. F1000Research, 10.7490/f1000research.1111735.1 (2016).
Li, R. et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25, 1966–1967, 10.1093/bioinformatics/btp336 (2009).
Kopylova, E., Noe, L. & Touzet, H. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28, 3211–3217, 10.1093/bioinformatics/bts611 (2012).
Noguchi, H., Park, J. & Takagi, T. MetaGene: prokaryotic gene finding from environmental genome shotgun sequences. Nucleic acids research 34, 5623–5630, 10.1093/nar/gkl723 (2006).
Testa, A. C., Oliver, R. P. & Hane, J. K. OcculterCut: A Comprehensive Survey of AT-Rich Regions in Fungal Genomes. Genome biology and evolution 8, 2044–2064, 10.1093/gbe/evw121 (2016).
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212, 10.1093/bioinformatics/btv351 (2015).
Data Citations
European Nucleotide Archive PRJEB24468 (2018)
European Nucleotide Archive PRJEB24469 (2018)
European Nucleotide Archive PRJEB24467 (2018)
European Nucleotide Archive PRJEB21682 (2018)
Dutreux, F. et al. figshare https://doi.org/10.6084/m9.figshare.c.4124543 (2018)
Acknowledgements
The authors are grateful to Oxford Nanopore Technologies Ltd for providing early access to the MinION device through the MinION Access Programme, and we thank the staff of Oxford Nanopore Technology Ltd for technical help. BI, CC, PW, and JMA are part of the MinION Access Programme (MAP) and JMA received travel and accommodation expenses to speak at Oxford Nanopore Technologies conferences. This work was supported by the Genoscope, the Commissariat à l’Energie Atomique et aux Energies Alternatives (CEA) and France Génomique (ANR-10-INBS-09-08). F. Dutreux was funded by a grant from PROMOSOL (Association pour la promotion de la sélection des plantes oléagineuses). The genetic map was obtained through a funding of the French National research agency (ANR project ANR-12-ADAP-0009 ‘Gandalf’).
Author information
Authors and Affiliations
Contributions
C.C. and A.L. optimized and performed the sequencing. J.L. prepared all RNA samples. M.H.B. supervised the production of RNA and DNA samples and performed the genetic map. F.D., C.D.S., B.I., A.C., N.L., E.J.G., L.A. and J.M.A. performed the bioinformatic analyses. T.R. and M.H.B. conceived the Leptolife project and led the Promosol project. F.D. and J.M.A. wrote the article. P.W. and J.M.A. supervised the study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
ISA-Tab metadata
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files made available in this article.
About this article

Cite this article
Dutreux, F., Da Silva, C., d’Agata, L. et al. De novo assembly and annotation of three Leptosphaeria genomes using Oxford Nanopore MinION sequencing. Sci Data 5, 180235 (2018). https://doi.org/10.1038/sdata.2018.235
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/sdata.2018.235
This article is cited by
-
Location and timing govern tripartite interactions of fungal phytopathogens and host in the stem canker species complex
BMC Biology (2023)
-
CusProSe: a customizable protein annotation software with an application to the prediction of fungal secondary metabolism genes
Scientific Reports (2023)
-
Three new pathogenicity genes in Leptosphaeria maculans identified by Agrobacterium-mediated insertional mutagenesis
Australasian Plant Pathology (2023)
-
Large-scale transcriptomics to dissect 2 years of the life of a fungal phytopathogen interacting with its host plant
BMC Biology (2021)
-
An automated and combinative method for the predictive ranking of candidate effector proteins of fungal plant pathogens
Scientific Reports (2021)
