
Abstract
Sarcophaga peregrina (flesh fly) is a frequently found fly species in Palaearctic, Oriental, and Australasian regions that can be used to estimate minimal postmortem intervals important for forensic investigations. Despite its forensic importance, the genome information of S. peregrina has not been fully described. Therefore, we generated a comprehensive gene expression dataset using RNA sequencing and carried out de novo assembly to characterize the S. peregrina transcriptome. We obtained precise sequence information for RNA transcripts using two different methods. Based on primary sequence information, we identified sets of assembled unigenes and predicted coding sequences. Functional annotation of the aligned unigenes was performed using the UniProt, Gene Ontology, and Kyoto Encyclopedia of Genes and Genomes databases. As a result, 26,580,352 and 83,221 raw reads were obtained using the Illumina MiSeq and Pacbio RS II Iso-Seq sequencing applications, respectively. From these reads, 55,730 contigs were successfully annotated. The present study provides the resulting genome information of S. peregrina, which is valuable for forensic applications.
Design Type(s) | transcription profiling by high throughput sequencing design • sequence assembly objective • data annotation objective |
Measurement Type(s) | transcription profiling assay |
Technology Type(s) | RNA sequencing |
Factor Type(s) | |
Sample Characteristic(s) | Sarcophaga peregrina • whole body |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others

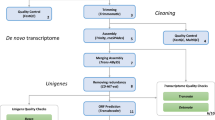
Background & Summary
Forensic entomology is an invaluable tool for estimating minimal postmortem interval (PMI) in criminal investigations. For the precise estimation of PMI, it is necessary to identify species and developmental stages of insects in corpses1–3. Some arthropods, particularly those belonging to the order Diptera (flies), are attracted to the bodies of dead animals. Flesh flies from the Sarcophagidae family usually appear on corpses slightly later than blow flies from the Calliphoridae family and are considered the second-most important species for forensic applications4–7. Flesh fly larvae are usually larger than Calliphoridae larvae at the same developmental stage, making them more conspicuous at the death scene and easier to sample by investigators8. The minimal PMI is usually estimated based on the developmental stage of the oldest larvae by calculating accumulated degree hours (ADH)8.
Sarcophaga peregrina (S. peregrina) is widely distributed in Palaearctic, Oriental, and Australasian regions9. It is found in many insect succession studies and at many death scenes10–14. In a previous study comparing insect succession patterns in human cadavers and animal carcasses, S. peregrina was found on human, pig, and rabbit corpses13. Importantly, S. peregrina was found in 7 out of 35 (20%) medicolegal autopsy cases in the northeastern area of Seoul, Korea15 and in 16 out of 42 (38%) autopsy cases in Saitama Prefecture, Japan16. S. peregrina is also related to parasitic diseases such as myiasis in both human and livestock, which is often forensically important as it can imply abuse17,18. Despite its forensic and medical significance, genomic information for S. peregrina has not yet been described.
In the present study, we carried out de novo transcriptome assembly in S. peregrina using high-throughput RNA sequencing (RNA-seq) followed by bioinformatic gene modeling. The assembled contigs were annotated, thereby enabling generation of the first gene catalogs for S. peregrina. The catalog of annotated S. peregrina genes in this study improves upon currently available dipteran transcriptome datasets and is the first comprehensive analysis of gene expression profiles for S. peregrina. Our data could be applied to RNA-based studies on this forensically important fly species including molecular approaches to the growth and development of S. peregrina for forensic investigations. Overall experimental workflow is summarized in Fig. 1.

Samples were prepared by pooling equal amounts of RNA from each developmental stage of S. peregrina, including early-, middle-, and late-instar larvae; middle-stage pupae; and adults. cDNA was synthesized and sequenced using the Illumina MiSeq platform with paired-end reads. For more accurate gene prediction of S. peregrina, we also employed the Pacbio RS II Iso-Seq system for full-length transcript sequencing. Analysis started with the assembly of full-length transcripts and corresponding isoforms using the de novo assembly program CLC Assembly Cell without a reference genome. The sequenced transcripts derived from Iso-Seq and MiSeq were combined, and the CD-HIT-EST program was used to construct the final standard transcript to eliminate redundancy. To examine the completeness of S. peregrina unigenes, we used the TransDecoder program BUSCO analysis and continued with functional analysis using the BLAST program. Quality control assessments were performed at each step.
Methods
Sample collection
We established a breeding colony from wild-type S. peregrina flies collected in the Northeastern region of Seoul, Korea. Flies were kept in a temperature-controlled chamber [24±0.8°C, 70±5% relative humidity, and 16:8 h (light:dark) photoperiod]. Specimens were prepared from F3 progeny at the following five developmental stages: early- (n = 30), middle- (n = 5), and late-instar larvae (n = 3); middle-stage pupae (n = 3); and newly-emerged adults (2 males and 2 females). We confirmed species identities of our specimens using nucleotide sequences of mitochondrial cytochrome c oxidase subunit I (COI) as described in our previous study15. Developmental times were quantified as a sequence of days and ADH using a developmental threshold temperature of 10.9°C19,20. A total ADH value spanning all five stages from egg to adult was calculated as 5,895 h. The collected larvae were observed under an Olympus SZX10 stereomicroscope (Olympus, Japan) to determine the larval instar based on the number of clefts in the posterior spiracle. The pupal stage was observed at a 4- or 8-h interval until adult eclosion. The wandering duration, pupation, and eclosion time points were recorded during the experiment. Whole body samples were quickly frozen in liquid nitrogen and stored at −70 °C for subsequent RNA extraction.
RNA preparation
Each sample was homogenized with liquid nitrogen in a mortar. Total RNA was extracted using a RNeasy mini kit (Qiagen GmbH, Hilden, Germany) according to the manufacturer’s instructions. Equal amounts of RNA samples from different developmental stages (10 μg per sample) were pooled for RNA sequencing. RNA concentration was assessed using a NanoDrop™ spectrophotometer (Thermo Fisher Scientific, Waltham, USA), and RNA integrity number values were calculated by an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, USA).
Library construction and sequencing
We purified poly(A) mRNA using Oligo (dT) magnetic beads (Qiagen), and the purified mRNA was broken into short fragments. Double-stranded cDNA was synthesized with sequencing adapters using the TruSeqTM Stranded mRNA prep Kit (Illumina). Finally, library sequence data were acquired using paired-end sequencing via the Illumina MiSeq platform. For more accurate gene prediction of S. peregrina, Iso-Seq using the Pacbio RS II system was employed for full-length transcript sequencing. Library construction and sequencing processing were conducted following the manufacturer’s instructions. Raw reads from MiSeq paired-end sequencing underwent pre-processing by removing Illumina TruSeqTM adapter sequences and low-quality sequences (<Q20) using trimmomatic21 with default parameters. To identify contaminant sequences for removal, clean reads without adapter and low-quality bases were mapped to bacterial and ocean metagenome databases downloaded from NCBI (ftp://ftp.ncbi.nlm.nih.gov/genomes/Bacteria, ftp://ftp.ncbi.nlm.nih.gov/genomes/Bacteria_DRAFT, ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/204/965/GCA_000204965.1_ASM20496v1) using the default setting of bowtie2. Those regions that did not map to the databases were subsequently removed from further analysis.
De novo assembly and dataset annotation
High-quality sequences were used in the subsequent assembly. Transcripts were assembled using CLC Assembly Cell (version 5.0; CLC bio, Waltham, MA, USA), which is optimized to present the best de novo results compared with a variety of assemblers that utilize MiSeq paired-end reads. Transcripts derived from Iso-Seq and Miseq were combined, and the CD-HIT-EST program (version 4.6.5)22,23 was used to construct the final standard transcript with default parameters (similarity 95%) to eliminate transcript redundancy. The coding region prediction of assembled transcripts was performed using TransDecoder (version 3.0.0; implemented in Trinity software) (http://transdecoder.github.io). We then assessed its overall inclusiveness/completeness by performing a benchmarking universal single-copy orthologs (BUSCO) analysis (https://busco.ezlab.org/)24. We used OrthoDB database of orthologs (www.orthodb.org) to define BUSCO sets for three major phylogenetic clades, which use 1,066 for Arthropoda, 2,799 for Diptera and 1,658 for Insecta near-universal single-copy orthologs. The Blast2GO program (E-value < 1e-3) was used to annotate the unigenes based on UniProt (http://www.uniprot.org/help/uniref) and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases (http://www.genome.jp/kegg/). Gene Ontology (GO) terms were assigned to each unigene based on the GO terms annotated to its corresponding homologs in the UniProt database. The obtained contigs of S. peregrina were then analyzed by gene family identification for annotation quality control. The following pipeline was used to cluster individual genes into gene families: (i) S. peregrina-to-D. melanogaster blastp was used to align all protein sequences with an E-value of 1e-3, and (ii) the gene families were clustered using OrthoMCL software25. Protein sequences of D. melanogaster were downloaded from Ensembl (release 85).
Data Records
Three types of datasets were generated in this study. The first dataset consists of RNA-seq raw reads of S. peregrina, which were submitted to the NCBI database (Data Citation 1 and Table 1). The second dataset contains the unigenes of S. peregrina (Data Citation 2 and Table 2). The third dataset comprises the annotation results for all databases and the predicted coding regions (CDSs) and protein information (Data Citation 3 and Table 3). This dataset also contains OrthoMCL results from the complete proteome of S. peregrina and D. melanogaster for examining transcript annotation completeness (Data Citation 3 and Table 3).
Technical Validation
Sequencing quality control
We evaluated sequencing quality to determine whether our results sufficiently cover the transcriptome of S. peregrina. A total of 26,580,352 raw reads were obtained by Illumina MiSeq platform paired-end sequencing. However, this conventionally applied short-read sequencing platform does not reliably distinguish many transcript isoforms. For more accurate gene prediction, we additionally employed Pacbio RS II Iso-Seq for full-length transcript sequencing. A total of 83,221 raw reads were obtained by full-length transcript sequencing (Table 4). We also tested samples using FastQC26 for Q20 and GC content (Table 4).
Assembly quality control
Trimmomatic21 was used to improve the overall quality of the assembly by removing adaptor contamination and serving as a quality control assessment tool for raw reads. Clean reads derived from bacterial and viral genomes were mapped to the bacterial and the ocean metagenome databases downloaded from NCBI by applying bowtie2. De novo assembly of the clean reads was performed using CLC Assembly Cell, which reconstructs full-length transcripts and corresponding isoforms without a reference genome. Transcripts derived from Iso-Seq and MiSeq were combined, and the CD-HIT-EST program was used to construct the final standard transcript with default parameters (similarity 95%) to eliminate transcript redundancy. After assembly and combination, 77,089 contigs were obtained. These contigs contain many isoforms. Coding-region prediction in the assembled transcripts was performed using the TransDecoder program implemented in Trinity software to examine the completeness of S. peregrina unigenes. As a result, 55,730 unigenes were identified by open reading frame prediction in the entire transcriptome of S. peregrina. The contig lengths of S. peregrina ranged from 237 to 13,704 bp with an average length of 623.43 bp (Table 5). The S. peregrina assemblies were also evaluated using the BUSCO arthropod, Diptera and Insecta gene sets, which use 1,066, 2,799 and 1,658 near-universal single-copy orthologs to assess the relative completeness of genome and transcriptome assemblies. The percentage of conserved genes identified in the S. peregrina assembly compares favorably with metrics reported for a number of insect transcriptomes and model insect genome assemblies (Table 6).
Annotation quality control
We estimated functional annotation results based on the aforementioned database and detailed information from the UniProt database (Table 7), which revealed that 33,991 unigenes (60.99%) were aligned to the UniProt database. The E-value distribution of the top hits showed that 39.22% of the sequences have strong homology (smaller than 1e-60) (Fig. 2a). Also, the top hit species distribution of matches with known sequences indicates that the majority of S. peregrina sequences show the highest homology with Musca domestica sequences (32.44%). The other most-represented species include insects like flies (Fig. 2b). All alignments were carried out using E-value thresholds of <1e-3. Next, Blast2GO was used to assign GO terms and functionally categorize the assembled S. peregrina contigs. Many of the assembled contigs correspond to at least one GO term (23,269 contigs, 41.75% of all contigs; Fig. 3). The annotated transcript sequences represent a significant contribution to the genomic information available for this species.

(a) E-value distribution of the top BLAST hits for each contig (E-value<1.0 e-3). (b) Hit species distribution.

Results are summarized in three main categories: biological process, cellular component, and molecular function. The y-axis indicates the number of contigs.
To identify the biological pathways active in S. peregrina, we mapped the 55,730 annotated sequences to reference canonical pathways in KEGG. A total of 6,335 unigenes (11.36%) were assigned to 132 known metabolic or signaling KEGG pathways (Table 7). Next, we applied OrthoMCL to the complete proteome of S. peregrina and D. melanogaster. From the dataset of 86,092 proteins (30,362 from D. melanogaster and 55,730 from S. peregrina), OrthoMCL categorized 37,365 proteins (14,584 for S. peregrina and 22,670 for D. melanogaster) into 8,378 groups (E-value < 1e-3). These results show that orthologs between two species can be identified using our data. There were 1.74 and 2.70 orthologs in the average ortholog group of S. peregrina and D. melanogaster (Table 8).
Usage Notes
The data provided in these experimental datasets can be used for two purposes. First, the raw reads can be used to conduct new analyses using different methods. Second, each analysis step can be repeated with specific technical changes, as all technical and experimental information is publicly available.
De novo assembly
The de novo assembly of RNA sequencing reads without a reference genome remains a challenge despite the development of many bioinformatic tools for data assembly and analysis27–30. In this study, a reference transcriptome for S. peregrina was sequenced and annotated using Illumina MiSeq and PacBio Iso-Seq sequencing technologies obtaining 7872.2 Mbp and 191.3 Mbp of transcriptome data, respectively, which further assembled into 55,730 unigenes. To the best of our knowledge, this is the first study to obtain whole transcriptome information using RNA sequencing in S. peregrina. In the past few years, interest in the forensic investigation of S. peregrina has increased in many countries. The limited genetic information and lack of understanding of the molecular mechanisms pertaining to S. peregrina development and reproduction have been the main obstacles preventing further studies. Our study provides the most extensive sequencing resource and genetic information of S. peregrina available to date, which provides a foundation for further molecular studies. Whole transcriptome data of S. peregrina were obtained using high-throughput sequencing, and unigenes were annotated using three main databases. Taken together, these results provide a solid foundation for further research on S. peregrina at the molecular level and can serve as an important genomic tool for forensic entomology communities.
Downstream analysis
The present study can be utilized as a reference for a variety of forensic entomological studies requiring RNA sequence information of S. peregrina and related species. For example, considering that S. peregrina belongs to necrophagous flies developing on corpses, identification of developmental stage-specific gene transcripts may greatly contribute to entomological approaches to PMI estimation. Determining the age of juvenile necrophagous flies is one of the key tasks in forensic entomology to provide evidence for the minimal PMI. As the age determination of the necrophagous fly larvae has largely relied on using morphological parameters, it is quite difficult to estimate the progressing pupal stage, which lasts about half of the total juvenile development without apparent morphological changes. In this regard, several previous studies have tried to identify sets of genes exhibiting a developmental stage-specific expression profiles in flesh flies by differential display31 or subtractive hybridization methods32. However, more systemic and genome-wide analyses on development-related gene expression in the flesh flies have not been yet carried out primarily due to lack of sufficient genomic information. The genome-wide transcriptome information and subsequent examination of development-associated expression profiles will provide novel molecular biomarkers for forensic applications of the S. peregrina. Identification of RNA species with highly developmental stage-specific expression profiles, in particular for pupal stages will greatly contribute to rapid and precise determination of the developmental stages, thereby estimation of the minimal PMI. To break new ground in the field of age determination of forensically relevant flies, additional transcriptome analysis would be required for each developmental stage. Data obtained in this study will serve as a basis to establish molecular age determination techniques based on S. peregrina and other necrophagous flies.
Additional information
How to cite this article: Kim. J. Y. et al. Comprehensive transcriptome analysis of Sarcophaga peregrina, a forensically important fly species. Sci. Data. 5:180220 doi: 10.1038/sdata.2018.220 (2018).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
References
Kiyoshi, S., Masataka, T. & Yasuhiro, A. Species identification of the forensically important flies in Iwate prefecture, Japan based on mitochondrial cytochrome oxidase gene subunit I (COI) sequences. Legal Medicine. 7, 175–178 (2005).
Amendt, J. et al. Best practice in forensic entomology-standards and guidelines. Int. J. Legal Med. 121, 90–104 (2007).
Brown, K., Thorne, A. & Harvey, M. Calliphora vicina (Diptera: Calliphoridae) pupae: A timeline of external morphological development and a new age and PMI estimation tool. Int. J. Legal Med. 129, 835–850 (2015).
Rodriguez, W. C. & Bass, W. M. Insect activity and its relationship to decay rates of human cadavers in East Tennessee. J. Forensic Sci. 28, 423–432 (1983).
Smith, K. G. V. A manual of forensic entomology. (Cornell University Pres, p99–p102, 1986).
Byrd, J. H. & Butler, J. F. Effects of temperature on Sarcophaga haemorrhoidalis (Diptera: Sarcophagidae) development. J. Med. Entomol 35, 694–698 (1998).
Szpila, K., Madra, A., Jarmusz, M. & Matuszewski, S. Flesh flies (Diptera: Sarcophagidae) colonising large carcasses in Central Europe. Parasitol. Res. 114, 2341–2348 (2015).
Grassberger, M. & Reiter, C. Effect of temperature on Lucilia sericata (Diptera: Calliphoridae) development with special reference to the isomegalen- and isomorphen-diagram. Forensic Sci. Int. 120, 32–36 (2001).
Xue, W., Verves, Y. G. & Du, J. A review of subtribe Boettcheriscina Verves 1990 (Diptera: Sarcophagidae), with descriptions of a new species and genus from China. Ann. Soc. Ent. Fr 47, 303–329 (2011).
Shi, Y., Liu, X., Wang, H. & Zhang, R. Seasonality of insect succession on exposed rabbit carrion in Guangzhou, China. Insect Sci 16, 425–439 (2009).
Guo, Y. D. et al. Identification of the forensically important sarcophagid flies Boerttcherisca peregrina, Parasarcophaga albiceps and Parasarcophaga dux (Diptera: Sarcophagidae) based on COII gene in China. Trop. Biomed. 27, 451–460 (2010).
Sukontason, K., Bunchu, N., Chaiwong, T., Moophayak, K. & Sukontason, K. L. Forensically important flesh fly species in Thailand: Morphology and developmental rate. Parasitol. Res. 106, 1055–1064 (2010).
Wang, Y. et al. Insect succession on remains of human and animals in Shenzhen, China. Forensic Sci. Int. 271, 75–86 (2017).
Shi, Y. W., Liu, X. S., Wang, H. Y. & Zhang, R. J. Seasonality of insect succession on exposed rabbit carrion in Guangzhou, China. Insect Science. 16, 425–439 (2009).
Shin, S. E. et al. The First survey of forensically important entomofauna collected from medicolegal autopsies in South Korea. Biomed Res Int 2015, 606728 (2015).
Toukairin, Y., Arai, T., Hoshi, T., Oliva Trejo, J. A. & Nogami, M. The geographical distribution of fly larvae on corpses in Saitama Prefecture in Japan during the summer season. Leg Med (Tokyo) 24, 75–77 (2016).
Fan, Z. D. . The key of common flies of China. 2nd edn, (Science Press, p689–p690 1992).
Chigusa, Y., Kirinoki, M. & Matsuda, H. Nosocomial myiasis due to Sarcophaga peregrina in an intensive care unit (ICU) in Japan. Med. Entomol. Zool 56, 355–358 (2005).
Cui, H. & Min, J. Forensic entomology. Chongqing Publishing House, p165 (2000).
Wang, Y., Wang, J. F., Zhang, Y. N., Tao, L. Y. & Wang, M. Forensically important Boettcherisca peregrina (Diptera: Sarcophagidae) in China: Development pattern and significance for estimating postmortem Interval. J. Med. Entomol 54, 1491–1497 (2017).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30, 2114–2120 (2014).
Li, W., Jaroszewski, L. & Godzik, A. Clustering of highly homologous sequences to reduce the size of large protein databases. Bioinformatics. 17, 282–283 (2001).
Li, W., Jaroszewski, L. & Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 22, 1658–1659 (2006).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 31, 3210–3212 (2015).
Li, L., Stoeckert, C. J. & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003).
Andrews, S. FASTQC: a quality control tool for high throughput sequence data. Babraham Bioinformaticshttp://www.bioinformatics.babraham.ac.uk/projects/fastqc (2010).
Zerbino, D. R. & Birney, E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829 (2008).
Simpson, J. T. et al. ABySS: a parallel assembler for short read sequence data. Genome Res. 19, 1117–1123 (2009).
Papanicolaou, A., Stierli, R., Ffrench-Constant, R. H. & Heckel, D. G. Next generation transcriptomes for next generation genomes using est2assembly. BMC Bioinformatics. 10, 447–453 (2009).
Martin, J. A. & Wang, Z. Next-generation transcriptome assembly. Nat. Rev. Genet. 12, 671–682 (2011).
Flannagan, R. D., Tammariello, S. P., Joplin, K. H., Cikra-Ireland, R. A., Yocum, G. D. & Denlinger, D. L. Diapause-specific gene expression in pupae of the flesh fly Sarcophaga crassipalpis. Proc Natl Acad Sci (USA) 95, 5610–5620 (1998).
Yamamoto-Kihara, M. & Kotani, E. Isolation and characterization of a C-type lectin cDNA specifically expressed in the tip of mouthparts of the flesh fly Sarcophaga peregrina. Insect Mol Biol 13, 133–140 (2004).
Data Citations
NCBI Sequence Read Archive SRP124481 (2017)
GenBank GGEP00000000 (2018)
Kim, J. Y. et al. figshare https://doi.org/10.6084/m9.figshare.c.4055423 (2018)
Acknowledgements
This work was supported by the Korean National Police Agency through the Research and Development of Police Science and Technology project (PA-G000001/2016-401). G.H.S., S.H.P., and S.K.K. were supported by the Korea University grant program. J.Y.K. and H.K.C. were supported by the Brain Korea 21 PLUS program.
Author information
Authors and Affiliations
Contributions
G.H.S. and S.H.P. conceived of and designed the study. J.Y.K., H.Y.L., S.E.S., H.K.C., and S.K.K. carried out experiments. J.Y.K., S.H.P., and G.H.S. analyzed data and wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
ISA-Tab metadata
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files made available in this article.
About this article

Cite this article
Kim, J., Lim, H., Shin, S. et al. Comprehensive transcriptome analysis of Sarcophaga peregrina, a forensically important fly species. Sci Data 5, 180220 (2018). https://doi.org/10.1038/sdata.2018.220
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/sdata.2018.220
