
Abstract
Recent advances in understanding the ecology of marine systems have been greatly facilitated by the growing availability of metagenomic data, which provide information on the identity, diversity and functional potential of the microbial community in a particular place and time. Here we present a dataset comprising over 5 terabases of metagenomic data from 610 samples spanning diverse regions of the Atlantic and Pacific Oceans. One set of metagenomes, collected on GEOTRACES cruises, captures large geographic transects at multiple depths per station. The second set represents two years of time-series data, collected at roughly monthly intervals from 3 depths at two long-term ocean sampling sites, Station ALOHA and BATS. These metagenomes contain genomic information from a diverse range of bacteria, archaea, eukaryotes and viruses. The data’s utility is strengthened by the availability of extensive physical, chemical, and biological measurements associated with each sample. We expect that these metagenomes will facilitate a wide range of comparative studies that seek to illuminate new aspects of marine microbial ecosystems.
Design Type(s) | biodiversity assessment objective • species comparison design |
Measurement Type(s) | DNA sequencing |
Technology Type(s) | high throughput sample analysis |
Factor Type(s) | geographic location • temporal_instant |
Sample Characteristic(s) | marine metagenome • Atlantic Ocean • Pacific Ocean • Pacific Ocean Gyre • Sargasso Sea • ocean biome |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others
Background & Summary
Microbial communities are key drivers of marine biogeochemistry. Our understanding of the incredible complexity and diversity of natural microbial populations has been greatly enhanced by the advent of cultivation-independent techniques for sequencing DNA directly from an environmental sample. Despite progress in describing the complexity of these natural systems, many gaps remain in our understanding of the distribution of genes and organisms in the oceans as well as the selective forces that structure community composition and distribution across space and time.
While previous large-scale marine sequencing efforts such as the Global Ocean Survey1,2 and Tara Oceans3 expeditions have greatly expanded our understanding of ocean microbiomes, these ecosystems remain vastly undersampled. The oceans present many challenges for sampling, including both their dynamic nature–e.g. weather, turbulence, movements of water masses, and mixing–as well as their remoteness. Further, understanding the forces that shape these communities requires detailed physical and chemical measurements associated with individual samples to provide information on the selective pressures that might play a role.
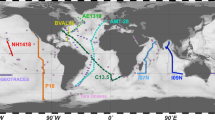
Here we present whole community metagenomic data from 610 samples collected in the Atlantic and Pacific Oceans. These data represent snapshots of microbial communities sampled across space and time, and are associated with physical and chemical measurements which are of value in addressing integrative research questions. The first set of metagenomes, collected under the auspices of the bioGEOTRACES component of the international GEOTRACES program4, comprises 480 samples collected in 2010-2011 (Data Citation 1). These samples come from 91 stations visited over four major cruise transects, with 2-10 depths (median 5) sampled at each station (Fig. 1; Table 1). An extensive suite of physical and chemical measurements, comprising over 147 unique data types including salinity, oxygen, temperature, nutrients, and detailed trace metal concentrations4, are available for these samples.

(a) Global map indicating the location of each sampling station where metagenomes were collected on the indicated cruise. Sample locations are shown in relation to sites sampled during two other large-scale marine metagenome sampling projects, the GOS and TARA datasets1–3 for context. (b) Depth distribution of metagenome samples along each of the four GEOTRACES cruises. Transect distances are calculated relative to the first station sampled in the indicated orientation. The depth distribution of samples collected below 250 m are not shown to scale for clarity (ranging from 281–5601 m; see Table 3 (available online only)).
The second set of metagenomes contains time series data collected at two long-term ocean study sites: Station ALOHA in the North Pacific Subtropical Gyre, sampled as part of the Hawai’i Ocean Time-series (HOT) program5, and the Bermuda-Atlantic Time-series Study (BATS) Station6 in the Sargasso Sea (Data Citation 2). Water samples were collected every month for two years (2003-2004) at both locations, and we sequenced libraries from 3 depths per month (between 1-180 m), representing surface water, the deep chlorophyll maximum, and the bottom of the euphotic zone (Fig. 2; Table 2). Two additional samples collected from each site in 2009 are also included (Table 3 (available online only)). This temporal sampling scheme provides opportunities to compare and contrast variations within and between these two oligotrophic ocean regimes across seasonal, inter-annual and intra-annual time scales. Station ALOHA, for example, remains stably stratified throughout much of the summer7, and is often considered to be a N-limited ecosystem8,9; BATS, on the other hand, is subject to deep winter mixing events and is generally considered to be a P-limited system6. Both HOT and BATS metagenomes are associated with concurrent measurements of numerous other parameters, including physical characterization (e.g., light, temperature, salinity), nutrient concentrations, biological process rate measurements, and in situ cell concentrations5,6.

Black dots indicate the time and depth of each sample sequenced from Station ALOHA (Hawai’i Ocean Time-Series; a, b) and the Bermuda-Atlantic Time Series station (BATS; c, d). Sampling scheme is depicted in the context of total bacterial counts (a, c) and chlorophyll abundance (b, d) data from HOT22 and BATS6. The middle depth samples were chosen to track the deep chlorophyll maximum.
The complete dataset contains over 5 terabases (in 1.67×1010 paired-end reads) of raw sequence data (Table 4) (Data Citation 1 and Data Citation 2). In addition to the paired-end reads, we also include a set of assembled contigs from each metagenome library (Data Citation 3 for GEOTRACES and Data Citation 4 for HOT and BATS). As these metagenomes represent the microbial community in whole water samples, sequences from bacteria (39% of reads), archaea (4%), eukaryotes (1%) and viruses (2%) are present in roughly the same proportions observed in other marine datasets10. Future improvements in reference databases will likely continue to reduce the number of unidentified reads and refine read recruitment.
We anticipate that these data will be useful for addressing a wide variety of research questions and generating new hypotheses across a broad range of disciplines including, but not limited to, microbial ecology, population genetics, evolution, and oceanography. In particular, the physical, chemical, and biological measurements associated with these samples enable studies of the relationships between microbial community structure, functional potential, biogeochemical cycles, and specific environmental variables.
Methods
Whole water samples were collected onto 0.2 μm filters and preserved using previously described protocols for qPCR sampling11–14. Briefly, water was transferred from the appropriate Niskin bottle into a clean 500 mL amber bottle which had been washed three times with seawater from the same Niskin bottle. Replicate filters were prepared from each water sample by passing 100 mL of seawater through a 25mm diameter, 0.2 μm pore size polycarbonate filter under vacuum (9 in Hg maximum pressure). Filters were then chased with 3 mL of sterile preservation solution (10 mM Tris, pH 8.0; 100 mM EDTA; 0.5 M NaCl) and then immediately transferred to cryovials and stored at -80 °C. All glassware and collection bottles were cleaned in 10% bleach followed by extensive rinsing with 18 mΩ water (Millipore Milli-Q).
Total community DNA was extracted using a phenol/chloroform-based extraction method15 that was slightly modified for these samples. Lysing Matrix E beads (MP Biomedicals), 400 ul Phenol:Chloroform:IAA (25:24:1) and 400 ul 2x TENS buffer (100 mM Tris-HCL pH 8.0, 40 mM EDTA, 200 mM NaCl, 2% SDS for 2x buffer) were added to a microcentrifuge tube containing the filter and then vigorously agitated using a beadbeater for 40 seconds. After spinning at 19,000 xg for 5 minutes, the aqueous phase was transferred into a Phase Lock Gel tube (5 Prime), mixed with an equal volume of chloroform, and then spun at ~27,000 x g for 5 minutes. The supernatant was removed and mixed with an equal volume of AMPure XP beads (Beckman Coulter), and incubated at room temperature for 10 minutes. Beads were washed twice with 75% ethanol, dried, and resuspended in 20 uL ultrapure glass distilled water (Teknova). Total DNA yield was quantified using the PicoGreen assay (ThermoFisher) with yields ranging from ~10-2600 ng total DNA.
Sequencing libraries were prepared and sequenced by the MIT BioMicro Center. Libraries were constructed using the NextEra XT kit (Illumina) on an automated Tecan Freedom EVO robotics platform, starting from 1ng of input DNA. Relevant adapter sequences for downstream quality trimming are 5’-CTGTCTCTTATACACATCTCCGAGCCCACGAGAC-3’ and 5’-CTGTCTCTTATACACATCTGACGCTGCCGACGA-3’. Target library insert length was ~250 nt. The resulting libraries were sequenced using the Illumina NextSeq platform to produce 150+150 nt paired reads. Sixteen metagenomes were multiplexed on each lane, and a median total of ~25 million raw paired-end reads was obtained for each sample (range: ~2.6–323 million, due to variations in library loading).
To characterize the overall taxonomic content of the metagenomes (see Background & Summary), low quality regions of sequencing data and Illumina adapter sequences were first removed using Trimmomatic (V0.36)16. The trimmed reads were then assigned taxonomy using Kaiju (V1.5.0)17 in MEM mode with the SEG low complexity filter enabled. Kaiju classification employed a database containing the NCBI nr database (consisting of 103 million bacterial, archaeal viral, fungal, and microbial eukaryotic protein sequences; accessed 2017-05-16)18, the Moore Foundation Marine Microbial Eukaryote Transcriptome Sequencing Project dataset19, and 729 marine single cell genomes20. Figures were generated using Ocean Data View (V4.7.10; http://odv.awi.de) and R (V3.3.2; https://www.R-project.org), with the assistance of a number of R-based tools (tidyverse: https://CRAN.R-project.org/package=tidyverse; sp: https://CRAN.R-project.org/package=sp; geosphere: https://CRAN.R-project.org/package=geosphere; patchwork: https://github.com/thomasp85/patchwork; ggworldmap: https://github.com/thackl/ggworldmap).
Metagenome assemblies of each library were generated using metaSPAdes21 (v3.9.0 and v3.10.1). Paired-end reads were first quality trimmed with Trimmomatic as above, and then used as input for the metaSPAdes algorithm (with the default --meta settings). Assembled contigs shorter than 200bp were discarded.
Code availability
No custom code was used to generate or process these data. Software versions and any relevant variables and parameters employed are as follows:
Trimmomatic (V0.36): -phred33 ILLUMINACLIP:NexteraPE-PE.fa:2:30:10
LEADING:3 TRAILING:3 SLIDINGWINDOW:10:20 MINLEN:75
Kaiju (V1.5.0): -a mem -x
SPAdes (V3.9.0): --meta
SPAdes (V3.10.1): --meta
Data Records
The raw Illumina sequencing reads and sets of assembled contigs for all metagenomes are available from the NCBI Sequence Read Archive (Data Citations 1, 2, 3, 4). Accession numbers, sample date/location, cruise information, and library size for each metagenome can be found in Table 3 (available online only).
Technical Validation
To confirm the reliability of the automated library preparation steps and ensure that sample cross-contamination was minimized, we randomly included blank buffer samples among our samples and verified that these did not yield successful libraries. Prior to sequencing, the quality of the Illumina libraries was assessed on a Fragment Analyzer (Advanced Analytical) to ensure that the median insert size and overall distribution was in the expected range (peak fragment length ~1200 bp, range 300-3000 bp), with a total yield >1 ng; libraries which did not meet these criteria were reprepared. Whenever possible, libraries with relatively low sequencing coverage (<1×107 paired-end reads) were subjected to additional rounds of sequencing. Sequencing quality (as assessed by per-base average sequence quality scores, quality over the length of the read, kmer overrepresentation, etc) was monitored by the MIT BioMicro Center’s automated sequencing analysis pipelines, and libraries were resequenced if necessary.
Usage Notes
All metagenomes are associated with standardized GEOTRACES, HOT, and BATS bottle identification numbers to enable cross-referencing with the relevant databases of physical, chemical, and biological measurements (Table 3 (available online only)). GEOTRACES data can be accessed from the British Oceanographic Data Centre (https://www.bodc.ac.uk/geotraces/). HOT data can be accessed from http://hahana.soest.hawaii.edu/hot/hot-dogs/index.html, and BATS data from http://bats.bios.edu/. GEOTRACES data are periodically updated, and users are encouraged to access the most recent data release. Note that each individual metagenome may not have associated measurements for all possible parameters. Access to data from all of these sources is subject to their respective data use policies.
The assembled contigs for each metagenome (Data Citations 3 and Data Citations 4) are deposited in the NCBI Sequence Read Archive as analysis objects with accession numbers as listed in Table 3 (available online only). Assemblies are also available from the iMicrobe database: Geotraces samples at https://www.imicrobe.us/#/projects/277; HOT samples at https://www.imicrobe.us/#/projects/271; and BATS samples at https://www.imicrobe.us/#/projects/276.
Some use cases for the metagenomes could benefit from overlapping the paired-end data to create longer reads for downstream analysis. In our experience, on average 51% of paired reads overlap. We also note that these reads and assembled contigs may contain a small amount of contamination arising from the sampling, library preparation, and/or sequencing steps. While we worked to minimize this as much as possible, note that the data described here have not been pre-screened.
Additional information
How to cite this article: Biller, S. J. et al. Marine microbial metagenomes sampled across space and time. Sci. Data 5:180176 doi: 10.1038/sdata.2018.176 (2018).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
References
Venter, J. C. et al. Environmental genome shotgun sequencing of the Sargasso Sea. Science 304, 66–74 (2004).
Rusch, D. B. et al. The Sorcerer II Global Ocean Sampling expedition: northwest Atlantic through eastern tropical Pacific. PLoS Biol 5, e77–431 (2007).
Pesant, S. et al. Open science resources for the discovery and analysis of Tara Oceans data. Scientific Data 2, 150023–16 (2015).
Anderson, R., Mawji, E., Cutter, G., Measures, C. & Jeandel, C. GEOTRACES: Changing the Way We Explore Ocean Chemistry. Oceanography 27, 50–61 (2014).
Karl, D. M. & Church, M. J. Microbial oceanography and the Hawaii Ocean Time-series programme. Nature Reviews Microbiology 12, 699–713 (2014).
Steinberg, D. K. et al. Overview of the US JGOFS Bermuda Atlantic Time-series Study (BATS): a decade-scale look at ocean biology and biogeochemistry. Deep-Sea Research Part II 48, 1405–1447 (2001).
Karl, D. M. & Church, M. J. Ecosystem Structure and Dynamics in the North Pacific Subtropical Gyre: New Views of an Old Ocean. Ecosystems 20, 433–457 (2017).
Wu, J., Sunda, W., Boyle, E. A. & Karl, D. M. Phosphate depletion in the western North Atlantic Ocean. Science 289, 759–762 (2000).
Van Mooy, B. A. S. & Devol, A. H. Assessing nutrient limitation of Prochlorococcus in the North Pacific subtropical gyre by using an RNA capture method. Limnology and Oceanography 53, 78–88 (2008).
Sunagawa, S. et al. Ocean plankton. Structure and function of the global ocean microbiome. Science 348, 1261359 (2015).
Ahlgren, N. A., Rocap, G. & Chisholm, S. W. Measurement of Prochlorococcus ecotypes using real-time polymerase chain reaction reveals different abundances of genotypes with similar light physiologies. Environmental Microbiology 8, 441–454 (2006).
Zinser, E. R. et al. Prochlorococcus ecotype abundances in the North Atlantic Ocean as revealed by an improved quantitative PCR method. Applied and Environmental Microbiology 72, 723–732 (2006).
Zinser, E. R., Coe, A., Karaca, E., Veneziano, D. & Chisholm, S. Influence of light and temperature on Prochlorococcus ecotype distributions in the Atlantic Ocean. Limnology and Oceanography 52, 2205–2220 (2007).
Malmstrom, R. R. et al. Temporal dynamics of Prochlorococcus ecotypes in the Atlantic and Pacific oceans. The ISME Journal 4, 1252–1264 (2010).
Urakawa, H., Martens-Habbena, W. & Stahl, D. A. High abundance of ammonia-oxidizing Archaea in coastal waters, determined using a modified DNA extraction method. Applied and Environmental Microbiology 76, 2129–2135 (2010).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Menzel, P., Ng, K. L. & Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nature Communications 7, 11257 (2016).
NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 46, D8–D13 (2018).
Keeling, P. J. et al. The Marine Microbial Eukaryote Transcriptome Sequencing Project (MMETSP): illuminating the functional diversity of eukaryotic life in the oceans through transcriptome sequencing. PLoS Biol 12, e1001889 (2014).
Berube, P. M. et al. Single cell genomes of Prochlorococcus, Synechococcus, and sympatric microbes from diverse marine environments. Scientific Data 5:180154 (2018).
Nurk, S., Meleshko, D., Korobeynikov, A. & Pevzner, P. A. metaSPAdes: a new versatile metagenomic assembler. Genome Res 27, 824–834 (2017).
Karl, D. M. & Lukas, R. The Hawaii Ocean Time-series (HOT) program: Background, rationale and field implementation. Deep-Sea Research Part II 43, 129–156 (1996).
Data Citations
NCBI Sequence Read Archive SRP110813 (2018)
NCBI Sequence Read Archive SRP109831 (2018)
NCBI BioProject PRJNA385854 (2018)
NCBI BioProject PRJNA385855 (2018)
Acknowledgements
This work was funded by grants from the Simons Foundation (Life Sciences Project Award ID 337262, S.W.C.; SCOPE Award ID 329108, S.W.C.), the Moore Foundation (Grant IDs GBMF495 and GBMF4511), and the National Science Foundation (OCE-1153588, OCE-1356460, and DBI-0424599 to S.W.C.). The authors wish to thank the GEOTRACES chief scientists for their support: Andrew Bowie (University of Tasmania), Philip Boyd (University of Tasmania), Edward Boyle (Massachusetts Institute of Technology), Gregory Cutter (Old Dominion University), Loes Gerringa (NIOZ Royal Netherlands Institute for Sea Research), Gideon Henderson (University of Oxford), William Jenkins (Woods Hole Oceanographic Institution), and Micha Rijkenberg (NIOZ Royal Netherlands Institute for Sea Research), as well as Gerhard Herndl (University of Vienna), James Moffett (University of Southern California) and Hein de Baar (NIOZ Royal Netherlands Institute for Sea Research) for organizational and logistical support of bioGEOTRACES. We thank the HOT and BATS cruise operations teams for their assistance with sampling, as well as the captains and crew of the R/V Kilo Moana, R/V Ka`imikai-O-Kanaloa, R/V Atlantic Explorer, R/V Pelagia, R/V Southern Surveyor, R/V Tangaroa, RRS James Cook, RRS Discovery, and R/V Knorr. We also thank the staff at the MIT BioMicro Center for their assistance with library preparation and sequencing. This paper is a contribution from the Simons Collaboration on Ocean Processes and Ecology (SCOPE) and the NSF Center for Microbial Oceanography: Research and Education (C-MORE).
Author information
Authors and Affiliations
Contributions
S.J.B. and P.M.B. managed the project and wrote the manuscript with contributions from all authors. K.D. and M.W. extracted DNA samples and coordinated sequencing. B.M.S., T.H., and S.L.H. contributed to dataset organization, quality control and preliminary analysis. T.H. assembled the metagenomes and assisted with generating figures. S.J.B., P.M.B., and S.W.C. designed the study. A.C. coordinated sample collection. K.B., H.B., T.J.B., D.D., C.H., D.H., J.E.J., E.W.M., T.R., E.S., and T.Y. executed the field sampling. S.W.C. supervised the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
ISA-Tab metadata
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files made available in this article.
About this article

Cite this article
Biller, S., Berube, P., Dooley, K. et al. Marine microbial metagenomes sampled across space and time. Sci Data 5, 180176 (2018). https://doi.org/10.1038/sdata.2018.176
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/sdata.2018.176
This article is cited by
-
Decoding populations in the ocean microbiome
Microbiome (2024)
-
Metagenomic exploration of Andaman region of the Indian Ocean
Scientific Reports (2024)
-
Genome-scale community modelling reveals conserved metabolic cross-feedings in epipelagic bacterioplankton communities
Nature Communications (2024)
-
Marine picoplankton metagenomes and MAGs from eleven vertical profiles obtained by the Malaspina Expedition
Scientific Data (2024)
-
Metagenome-assembled genomes reveal greatly expanded taxonomic and functional diversification of the abundant marine Roseobacter RCA cluster
Microbiome (2023)
