
Abstract
Interpreting the functional impact of noncoding variants is an ongoing challenge in the field of genome analysis. With most noncoding variants associated with complex traits and disease residing in regulatory regions, altered transcription factor (TF) binding has been proposed as a mechanism of action. It is therefore imperative to develop methods that predict the impact of noncoding variants at TF binding sites (TFBSs). Here, we describe the update of our MANTA database that stores: 1) TFBS predictions in the human genome, and 2) the potential impact on TF binding for all possible single nucleotide variants (SNVs) at these TFBSs. TFBSs were predicted by combining experimental ChIP-seq data from ReMap and computational position weight matrices (PWMs) derived from JASPAR. Impact of SNVs at these TFBSs was assessed by means of PWM scores computed on the alternate alleles. The updated database, MANTA2, provides the scientific community with a critical map of TFBSs and SNV impact scores to improve the interpretation of noncoding variants in the human genome.
Design Type(s) | data integration objective • database creation objective • transcription factor binding site prediction objective |
Measurement Type(s) | transcription factor binding site |
Technology Type(s) | prediction |
Factor Type(s) | cell line • antibody • transcription factor |
Sample Characteristic(s) | Homo sapiens |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others


Background & Summary
Understanding the relationship between DNA sequence variation (genotype) and observable traits and diseases (phenotype) is one of the central paradigms of the post-genomics era. While most analyses have focused on the ~2% of the genome that codes for proteins, genome-wide association studies have shown that up to 88% of disease- and trait-associated variants are located in the 98% of the genome that is noncoding1. Several computational tools, such as SIFT2 and Polyphen3, are well established for the assessment of the deleterious impact of coding variation on protein functions yet interpreting the functional impact of noncoding variants continues to be challenging4.
Recently, bioinformatics methods have been developed for scoring the impact of noncoding variants based on their pathogenicity and regulatory capacity (Table 1). These methods vary both in their algorithmic approaches and the underlying genomic features used. For instance, evolutionary conservation5 can be used to evaluate nucleotides under purifying selection, and experimental data such as histone modifications6, chromatin accessibility7,8, and DNA methylation9 are used to identify biochemically active DNA, which is indicative of regulatory capacity.
Transcription factors (TFs) are sequence-specific DNA-binding proteins that regulate gene transcription10. Genomic locations at which TFs interact with DNA are defined as TF binding sites (TFBSs). They are typically short (6–10 bp) and often exhibit degeneracy. Chromatin immunoprecipitation combined with sequencing (ChIP-seq)11 provides in vivo TF-DNA interactions at ~200–300 bp resolution. These ChIP-seq regions are expected to encompass the 6–10 bp fragments corresponding to TF-DNA interactions (TFBSs). The ReMap database12 is a publicly available resource providing an atlas of such regions obtained from 2,829 uniformly processed human ChIP-seq data sets.
The DNA sequences bound by a given TF can be represented as position frequency matrices (PFMs), which count the number of occurrences of each nucleotide within the TFBSs for that TF13. PFMs can be converted into probabilistic computational models, namely position weight matrices (PWMs), which can be used to predict TFBSs on any DNA sequence (reviewed by Wasserman and Sandelin14). Several databases of PFMs exist15, including the recently updated JASPAR database16, which stores manually-curated and non-redundant DNA-binding profiles such as PFMs for TFs across six taxonomic groups.
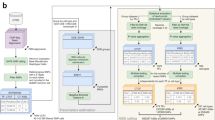
With most noncoding variants associated with complex traits and disease residing in regulatory sequences17, it is expected that some will alter the binding of TFs to DNA18,19. Therefore, it is imperative to develop methods that prioritize noncoding variants based on their impact on TF-DNA interactions. In 2015, we developed MANTA, a Mongo database for the analysis of TFBS alterations, to study the impact of regulatory mutations in B-cell lymphomas20. The database stores TFBSs in ChIP-seq regions predicted using PWMs derived from the JASPAR database, as well as the potential impact on TF binding of all possible single nucleotide variants (SNVs) that could occur at these TFBSs (Fig. 1). Building on the recent updates of both the JASPAR and ReMap databases, we have largely expanded MANTA. This second release of the database, MANTA2, hosts over 48 million TFBS predictions within ChIP-seq regions of 225 human TFs, covering about 8% of the human genome, together with computed impact scores for all possible overlapping SNVs. Hence, MANTA2 provides the scientific community with a critical map of TFBSs and SNV impact scores for the interpretation of noncoding variants in the human genome.

a) Intersection of the ReMap ChIP-seq regions with JASPAR TFBS predictions to produce a set of TFBSs with both experimental and computational evidence of TF binding. A mock example of JUN is given for a region on chromosome one. b) A matrix representing the difference in PWM score for all possible SNVs compared to the reference sequence at that TFBS, including negative impact (−), positive impact (+), and no change (0) of score. Black boxes indicate that nucleotides of the reference TFBS sequence are not stored in the database. The sequence logo for JUN is provided below the matrix where the information content is proportional to the size of the nucleotide letters. c) Mock distribution of TFBS SNV impact scores when considering all possible SNVs in the TFBS. The distribution is annotated with examples of decreased TF binding capacity (red), no change in TF binding capacity (yellow), and increased TF binding capacity (green).
Methods
Transcription factor binding site predictions
From ReMap12, we retrieved 1,902 uniformly processed ChIP-seq data sets (i.e. sets of ChIP-seq regions) for 227 human TFs for which we had binding profiles in JASPAR16. Each ChIP-seq data set was paired with one or more PFMs associated to the ChIP’ed TFs from the JASPAR CORE vertebrates collection (see Supplementary Table 1). For each pair, we intersected the ChIP-seq regions with the corresponding TFBSs predicted for the ChIP’ed TF using bedtools intersect21 with "-wa -wb" options to preserve the original coordinates. The PWM-based TFBS predictions are publicly available as part of the JASPAR human genome track at http://expdata.cmmt.ubc.ca/JASPAR/downloads/UCSC_tracks/2018/hg38/tsv/. The intersection resulted in 48,512,399 TFBSs for 225 TFs, covering 255,918,025 bp of the human genome (Fig. 1a). No overlap was found for 2 TFs between the ChIP-seq regions and PWM-based TFBS predictions. Note that all data relates to the build 38 of the Genome Reference Consortium human genome (hg38).
Computation of SNV impact scores
For each TFBS, we computed the impact on TF binding of all possible overlapping SNVs as described in the manuscript describing MANTA20 (Fig. 1b). First, both strands of the 2n−1 bp region centered around each possible SNV, where n is the length of the considered PWM, were scanned with the corresponding PWM using the TFBS Perl module22 (version 0.7.1) to identify the best PWM score on the alternate allele. Note that we only kept the best match per SNV. We then computed the distribution of PWM scores for all these SNVs and calculated the corresponding mean, m, and standard deviation, sd. For each SNV, the final impact score was calculated as the Z-score of its TFBS score, S, within the distribution of alternate PWM scores at that TFBS (i.e. (S−m)/sd). Users can refer to the webinar video describing the original MANTA database (http://www.cisreg.ca/Webinars/JASPAR_BioPython_MANTA.flv). Therefore, for each SNV, MANTA stores its associated reference and alternate TFBS PWM scores and locations, along with the computed impact score.
Validation using heterozygous TF-binding events
We downloaded ChIP-seq data for 35,703 TF-binding events at heterozygous sites in GM12878 and HeLa cells for 36 different TFs18. For each event, allelic imbalance was calculated as the number of ChIP-seq reads mapped on the alternate allele divided by the total number of reads mapped at that position (Fig. 2a). The coordinates from the original publication refer to the hg19 version of the human genome; we used the liftOver tool from the UCSC Genome Browser23 to convert them to the hg38 assembly (the conversion process failed for 12 coordinates).

a) Allelic imbalance is calculated as the number of ChIP-seq reads mapped on the alternate allele divided by the total number of reads mapped at heterozygous sites. b) MANTA2 impact scores correlate with allelic imbalance of ChIP-seq data. Events (blue dots) are plotted with respect to their allelic imbalance of ChIP-seq reads (x-axis) and impact scores from MANTA2 (y-axis). The Pearson coefficient (R) and P-value (p) of the correlation between allelic imbalance and impact score are provided in the plot.
Code availability
MANTA2 is freely distributed as a GitHub repository at https://github.com/wassermanlab/MANTA2.
Data Records
The Mongo database dump of MANTA2, is deposited as a tarball on Zenodo (Data Citation 1).
Technical Validation
The quality and technical validation of the ChIP-seq data and TFBS predictions is described in the 2018 manuscripts of ReMap12 and JASPAR16, respectively, and is summarised below.
ReMap ChIP-seq data
ReMap ChIP-seq datasets were uniformly processed using a well-established pipeline12. ChIP-seq reads were aligned to the human genome using bowtie2 (ref. 24) (version 2.2.9) using options “-end-to-end” and “-sensitive”. When necessary, reads were trimmed and polymerase chain reaction duplicates were removed from the alignments with samtools rmdup25. ChIP-seq regions were identified using the MACS2 peak-calling tool26 (version 2.1.1.2) with default parameters. The quality of all ChIP-seq datasets was assessed based on metrics developed by the ENCODE consortium27.
JASPAR TFBS predictions
JASPAR TFBSs were predicted by scanning the human genome using two different methods16: the TFBS Perl module22 (version 0.7.1) and FIMO28, as distributed within the MEME suite29 (version 4.11.2). FIMO is one of the best performing tools for scanning DNA sequences with PWMs to predict TFBSs30. To scan the human genome with the BioPerl TFBS module, PFMs were converted to PWMs and predictions with a relative score ≥0.8 were kept. In preparation for the FIMO scan, PFMs were reformatted to MEME motifs and motifs that matched with a P-value <0.05 were kept. For quality control, TFBS predictions that were not consistent between the two methods were filtered out. Such consistency ensures, for instance, technical validation for the coordinates of the TFBS predictions.
MANTA2
The technical validation of MANTA2 involved assessing data quality and database integrity controls. A spot check data quality control was performed using the UCSC Genome Browser23. For 15 randomly selected TFBSs (of different TFs) from MANTA2 we manually checked that: 1) the TFBS overlapped a ReMap ChIP-seq region associated with that TF; 2) the JASPAR PFM matched the start, end, and strand stored for that TFBSs; and 3) the stored SNVs for that TFBS had the expected impact on TF binding. Moreover, we assessed the usefulness of MANTA2 impact scores on an external dataset of heterozygous TF-binding events18. As expected, the allelic imbalance calculated for ChIP-seq reads (see Methods) significantly correlated with the impact scores from MANTA2 (Pearson correlation coefficient=0.567, P-value=3.7e-127; Fig. 2b). Additionally, we checked the database integrity for MANTA2 by dumping and restoring the database on common operating systems and workstations. Finally, we tested the command line and web interface access to MANTA2 (see Usage Notes section) to interpret variant files in VCF, GFF, and BED format.
Usage Notes
MANTA2 can be accessed either programmatically or via its web interface. To access the database programmatically, users must first clone (i.e. “git clone https://github.com/wassermanlab/MANTA2.git”) or download MANTA2 from GitHub (see Code availability in the Methods section). The script “search_manta2.py” provides programmatic access to MANTA2. It requires the following inputs:
-
The name of the MANTA2 database in the MongoDB system (option “-d”)
-
The name of the server where the MongoDB system is hosted (option “-H”)
-
A user with “read” privileges to the MANTA2 database (option “-u”)
-
The password for that user (option “-p”)
-
A file containing a list of variants in “VCF”, “BED” or “GFF” format (option “-i”)
Non-mandatory options include:
-
The format of the input variant file (option “-t”; by default the script tries to identify the input format automatically)
-
The name of a file to output the results (option “-o”; by default is set to the standard output stream (stdout))
As a usage example, the MANTA2 database hosted by the Wasserman lab can be accessed as follows: “python search_manta2.py -d manta2 -H manta.cmmt.ubc.ca -u manta_r -p mantapw -i <variant file>”.
A variant file can be obtained by executing the shell script: “bash ./examples/get_VCF_example.sh”.
The resulting VCF file (i.e. “chr20.vcf”) contains high-confidence SNP, small indel, and homozygous reference calls on chromosome 20 from the Genome in a Bottle (version 3.3.2) sample HG001 (ref. 31). In response, “search_manta2.py” returns all TFBS predictions potentially impacted by these variants as tab-separated values. For each TFBS alteration, the script provides the variant information along with the associated wild-type (reference) and mutated (alternative) TFBS information, including:
-
the chromosome and position of the variant;
-
the reference and alternative alleles at that genomic location;
-
the mutation ID (if the input file format allowed for it, otherwise the field is displayed as “.”);
-
the TF name and associated JASPAR profile ID;
-
the start, end and strand, as well as the absolute (raw) and relative scores for both the reference and alternative TFBSs;
-
and the impact score.
Users who plan on performing large numbers of searches should create a local build of the MANTA2 database. Instructions are provided in the “README.md” file of the GitHub repository.
The MANTA2 database hosted by the Wasserman lab can also be accessed via a dedicated web server at http://manta.cmmt.ubc.ca/manta2. Similar to the “search_manta2.py” script, the server requires as input a list of variants in VCF, GFF, or BED format (see help page), and it returns all TFBS predictions potentially impacted by these variants as a tab-separated values table. The table can be sorted on any column by clicking on the column header.
Additional information
How to cite this article: Fornes, O. et al. MANTA2, update of the Mongo database for the analysis of transcription factor binding site alterations. Sci. Data 5:180141 doi: 10.1038/sdata.2018.141 (2018).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
References
Maurano, M. T. et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195 (2012).
Ng, P. C. & Henikoff, S. Predicting Deleterious Amino Acid Substitutions. Genome Res. 11, 863–874 (2001).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010).
Mathelier, A., Shi, W. & Wasserman, W. W. Identification of altered cis-regulatory elements in human disease. Trends Genet. 31, 67–76 (2015).
Bejerano, G. Ultraconserved Elements in the Human Genome. Science 304, 1321–1325 (2004).
Tan, M. et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 146, 1016–1028 (2011).
Boyle, A. P. et al. High-resolution mapping and characterization of open chromatin across the genome. Cell 132, 311–322 (2008).
Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y. & Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013).
Varley, K. E. et al. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 23, 555–567 (2013).
Vaquerizas, J. M., Kummerfeld, S. K., Teichmann, S. A. & Luscombe, N. M. A census of human transcription factors: function, expression and evolution. Nat. Rev. Genet. 10, 252–263 (2009).
Johnson, D. S., Mortazavi, A., Myers, R. M. & Wold, B. Genome-Wide Mapping of in Vivo Protein-DNA Interactions. Science 316, 1497–1502 (2007).
Chèneby, J., Gheorghe, M., Artufel, M., Mathelier, A. & Ballester, B. ReMap 2018: an updated atlas of regulatory regions from an integrative analysis of DNA-binding ChIP-seq experiments. Nucleic Acids Res. 46, D267–D275 (2018).
Stormo, G. D. Modeling the specificity of protein-DNA interactions. Quantitative Biology 1, 115–130 (2013).
Wasserman, W. W. & Sandelin, A. Applied bioinformatics for the identification of regulatory elements. Nat. Rev. Genet. 5, 276–287 (2004).
Stormo, G. D. DNA Motif Databases and Their Uses. Curr. Protoc. Bioinformatics 51, 2.15.1–6 (2015).
Khan, A. et al. JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 46, D260–D266 (2018).
Albert, F. W. & Kruglyak, L. The role of regulatory variation in complex traits and disease. Nat. Rev. Genet. 16, 197–212 (2015).
Shi, W., Fornes, O., Mathelier, A. & Wasserman, W. W. Evaluating the impact of single nucleotide variants on transcription factor binding. Nucleic Acids Res. 44, 10106–10116 (2016).
Kumar, S., Ambrosini, G. & Bucher, P. SNP2TFBS - a database of regulatory SNPs affecting predicted transcription factor binding site affinity. Nucleic Acids Res. 45, D139–D144 (2017).
Mathelier, A. et al. Cis-regulatory somatic mutations and gene-expression alteration in B-cell lymphomas. Genome Biol. 16, 84 (2015).
Quinlan, A. R. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr. Protoc. Bioinformatics 47, 11.12.1–34 (2014).
Lenhard, B. & Wasserman, W. W. TFBS: Computational framework for transcription factor binding site analysis. Bioinformatics 18, 1135–1136 (2002).
Tyner, C. et al. The UCSC Genome Browser database: 2017 update. Nucleic Acids Res. 45, D626–D634 (2017).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Landt, S. G. et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 22, 1813–1831 (2012).
Grant, C. E., Bailey, T. L. & Noble, W. S. FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018 (2011).
Bailey, T. L. et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208 (2009).
Jayaram, N., Usvyat, D. & R. Martin, A. C. Evaluating tools for transcription factor binding site prediction. BMC Bioinformatics (2016); doi:10.1186/s12859-016-1298-9.
Zook, J. M. et al. Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls. Nat. Biotechnol. 32, 246–251 (2014).
Data Citations
Fornes, O., Gheorghe, M., Richmond, P. A., Arenillas, D. J., Wasserman, W. W., & Mathelier, A. Zenodo https://doi.org/10.5281/zenodo.1044747 (2017)
Acknowledgements
We thank Alice M. Kaye and Rachelle Farkas for proofreading the manuscript, and Georgios Magklaras and his team for IT support. We acknowledge the support provided by WestGrid (https://www.westgrid.ca/) and Compute Canada/Calcul Canada (https://www.computecanada.ca/). A.M. and M.G. were supported by funding from the Norwegian Research Council, Helse Sør-Øst, and the University of Oslo through the Centre for Molecular Medicine Norway (NCMM), which is part of the Nordic European Molecular Biology Laboratory Partnership for Molecular Medicine. D.J.A., O.F., P.A.R., and W.W.W. were supported by funding from Genome Canada and the Canadian Institutes of Health Research (OnTarget grants 255ONT and BOP-149430), the Natural Sciences and Engineering Research Council of Canada (discovery grant RGPIN-2017-06824), the Weston Brain Institute (20R74681), and the BC Children’s Hospital Foundation and Research Institute (UBC:17W33804 award to P.A.R.).
Author information
Authors and Affiliations
Contributions
A.M. and M.G. provided the ReMap ChIP-seq regions, and O.F. the JASPAR TFBS predictions. M.G. intersected the two data sets and A.M. generated the TFBS SNV scores. O.F. assessed the MANTA2 scores against heterozygous TF-binding events. D.J.A. updated MANTA and created the web server. P.A.R. generated the figures and tables, and reviewed the most relevant literature. A.M., O.F., and W.W.W. devised the project and wrote the manuscript with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Supplementary information accompanies this paper at
ISA-Tab metadata
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files made available in this article.
About this article

Cite this article
Fornes, O., Gheorghe, M., Richmond, P. et al. MANTA2, update of the Mongo database for the analysis of transcription factor binding site alterations. Sci Data 5, 180141 (2018). https://doi.org/10.1038/sdata.2018.141
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/sdata.2018.141
