
Abstract
Pseudorabies virus (PRV) is an alphaherpesvirus of swine. PRV has a large double-stranded DNA genome and, as the latest investigations have revealed, a very complex transcriptome. Here, we present a large RNA-Seq dataset, derived from both short- and long-read sequencing. The dataset contains 1.3 million 100 bp paired-end reads that were obtained from the Illumina random-primed libraries, as well as 10 million 50 bp single-end reads generated by the Illumina polyA-seq. The Pacific Biosciences RSII non-amplified method yielded 57,021 reads of inserts (ROIs) aligned to the viral genome, the amplified method resulted in 158,396 PRV-specific ROIs, while we obtained 12,555 ROIs using the Sequel platform. The Oxford Nanopore’s MinION device generated 44,006 reads using their regular cDNA-sequencing method, whereas 29,832 and 120,394 reads were produced by using the direct RNA-sequencing and the Cap-selection protocols, respectively. The raw reads were aligned to the PRV reference genome (KJ717942.1). Our provided dataset can be used to compare different sequencing approaches, library preparation methods, as well as for validation and testing bioinformatic pipelines.
Design Type(s) | transcription profiling by high throughput sequencing design • parallel group design |
Measurement Type(s) | messenger RNA • total RNA • Coding Region |
Technology Type(s) | RNA sequencing |
Factor Type(s) | temporal_interval • assay material selection • cap analysis of gene expression • enzymatic amplification • size fractionation • nucleic acid library construction protocol • Technology Platform |
Sample Characteristic(s) | Suid herpesvirus 1 strain Kaplan |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others

Background & Summary
Pseudorabies virus (PRV) is a causative agent of Aujeszky’s disease (AD)1 in pigs. PRV has a double-stranded DNA genome with a size of approximately 143 kbp. PRV is often employed in laboratories to study the molecular pathomechanism of the herpesviruses2. It is also a suitable tool as concerns gene and tumour therapy3, as well as for mapping of neuronal circuits4–8. This virus has also been used as live vaccines against AD9–11.
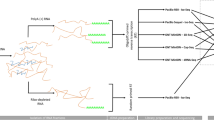
Here, we provide a large dataset derived from RNA-Seq experiments including different next-generation sequencing (NGS) – and third-generation sequencing (3rdGS) techniques (Fig. 1). Our aim with this study was to provide a dataset that can be used for comparison of the different sequencing platforms and library preparation methods using PRV as a model organism. In addition, these data are also applicable for identifying novel coding and non-coding transcripts, transcript isoforms, splice variants of PRV, and for defining full-length transcripts by using a combination of sequencing platforms.

One of the most popular NGS platforms, the Illumina HiScanSQ was used to generate high quality short-reads and extremely high coverage throughout the entire PRV genome. Random-primed cDNA library was prepared from viral RNAs. Paired-end RNA sequencing was carried out to characterize novel splice isoforms, as well as to obtain general information on the transcription activity of PRV12. PolyA-sequencing was used to determine the 3′-ends of RNA molecules. With this technique, we were able to detect alternative polyadenylation events in the PRV transcripts. Both libraries were run on a single flow cell resulting in 1.3 million 100 bp paired-end reads from the random hexamer-primed libraries, and 10 million 50 bp single-end reads from the poly(A)-enriched RNA-Seq, aligning to the viral reference13 (KJ717942.1).
Although the error rate of 3rdGS techniques is higher than those of NGS’s14, they are able to identify novel full-length transcripts15–17 and are therefore more applicable for global transcriptome profiling and RNA isoform detection compared to short-read techniques.
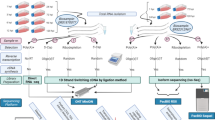
The Real-Time Sequencer II (RSII) and the Sequel 3rdGS platforms from Pacific Biosciences (PacBio) and the Oxford Nanopore Technologies (ONT) MinION 3rdGS device were used to characterize the static18,19 and dynamic20 PRV transcriptome. These sequencing techniques, with the library preparation methods [e.g. non-amplified SMRT method and amplified, Iso-seq protocol from the PacBio; full-length cDNA-sequencing, direct RNA-sequencing, and cDNA-sequencing on 5′Cap-selected samples from ONT, (Fig. 1,2)] used in these studies made it possible to identify several hundreds of novel transcript isoforms (including 3′- and 5′ UTR variants, and splice isoforms), as well as dozens of protein-coding and non-coding RNAs and numerous complex transcripts of PRV.

Seventy-one SMRT Cells were run on RSII system. P5-C3 chemistry and 180-minute data collection mode was used for the non-amplified samples, while P6-C4 enzymes were applied and 240 or 360 min movies were recorded for the amplified samples. cDNAs were sequenced on a single Sequel SMRT Cell with P6-C4 reagents; 10 h run-time was applied. Altogether seven MinION flow cells were used for the different ONT approaches.
The raw sequencing reads were mapped to the above-mentioned reference genome. Sequencing on the RSII platform resulted in 215,417 reads of inserts (ROIs), while the utilized nanopore sequencing methods generated altogether 194,232 PRV specific reads (Table 1). The average read lengths aligning to the PRV genome were 1,326 bp for PacBio RSII, 1,763 bp for the Sequel and 827 bp for ONT. It should be noted that the library preparation and size-selection methods resulted in different samples in length (Table 2).
This dataset can help explore the advantages and disadvantages associated with each sequencing method used in this work. This approach can be used for the analysis of multiple features of the sequencing platforms, including read length, base-calling error rate, coverage and mappability. The application of the various sequencing techniques can be evaluated by the analysis of the identified transcript isoforms, and the quantification of the transcriptome comparing the performance of Illumina, PacBio and ONT. This dataset is also useful for the analysis of the transcriptome complexity of PRV. Our data include a sub-dataset which can be used for the transcriptome analysis of PRV during an infection period including six different time-points.
Here we provide a detailed overview of the library preparation techniques and a description of the data (Table 3, Figs. 1 and 2).
Methods
Schematic overviews of the methodological workflow are shown in the flowchart of Figs. 1 and 2. The applied reagents and utilized approaches are listed in Table 4.
Cells, viruses and infection conditions
Immortalized porcine kidney-15 (PK-15; ATCC® CCL-33™) cells were used for the propagation of pseudorabies virus strain Kaplan (PRV-Ka) at 37 °C and 5% CO2 in Dulbecco’s modified Eagle medium (DMEM, Gibco Invitrogen) supplemented with 5% foetal bovine serum (FBS; Gibco Invitrogen). The virus stock was originally obtained from the Kaplan Lab (Department of Microbiology, Vanderbilt University School of Medicine, Nashville, Tennessee)21, but Vanderbilt University received it from Dr. Richard F. Haff in a suspension of infected mouse brain22. Gentamycin (80 μg/ml) was also added to the cell culture medium. The virus stock was prepared as follows: the medium was removed from the rapidly-growing semi-confluent PK-15 cells then it was infected with the Kaplan strain of PRV (a multiplicity of infection of 0.1 plaque-forming unit (pfu)/cell). Infected cells were incubated until complete cytopathic effect was observed. Samples were taken through three times freeze-thaw cycles, followed by centrifugation at 10,000 g for 15 min. The titre of the virus stock was determined in PK-15 cells. For all experiments, cells were infected with a high MOI (10 pfu/cell) and incubated for 1 h, followed by removal of the virus suspension and washing of the cells with phosphate-buffered saline (PBS). The number of cells in a culture flask was 5 × 106. After the addition of new medium to the cells, they were incubated for 1, 2, 4, 6, 8, 10, 12, 14, 16, 18, 20, 22 or 24 h pi and they both were mixed for Illumina sequencing (Table 3). One, 2, 4, 6, 8 and 12 h of incubation were used for the non-amplified PacBio sequencing, while the 1, 2, 4, 6 and 8 h pi samples were utilized for the PacBio amplified, Iso-Seq protocol. Samples from different time points were individually sequenced on the RSII, but they were also mixed for PacBio sequencing (Table 3). The incubation time was 1, 2, 3, 4, 5, 6, 7, 8, 9, 12, 18 and 24 h and a mixture from them was used for all types of ONT sequencing (Table 3).
RNA purification
Isolation of total RNAs The NucleoSpin® RNA II kit (Macherey-Nagel) was used to isolate RNA from samples for Illumina sequencing, while the new version, the NucleoSpin® RNA kit (Macherey-Nagel) was used for all the other samples, as was described earlier12,18,20. Briefly, cells were collected by centrifugation and lysed by incubation in a solution containing large amounts of chaotropic ions. This buffer inactivates the RNase. Nucleic acid molecules bind to the silica membrane. All samples were handled with DNase I solution (provided by the kit) to remove residual DNA contaminations. Total RNAs were eluted from the membrane in RNase-free water. To eliminate the potential remaining DNA contamination, samples were treated by Ambion® TURBO DNA-free™ Kit. The final concentrations of the RNA samples were determined by Qubit®. 2.0 Fluorometer using Qubit RNA BR Assay Kit (Life Technologies). RNA quality was assessed with the Agilent Bioanalyzer 2100 and RIN scores above 9.6 were used for cDNA production. RNA samples were stored at −80 °C until further use. Samples were made as follows: The Illumina oligodT- and random-primed sequencing reactions were carried out from the same RNA mixture. The libraries for the kinetic analysis and for the mixed sequencing using PacBio RSII (non-amplified method) were all prepared from different cell culture flasks (containing 5×106 cells/flask), but the same virus stock was used for these infections. For the amplified RSII sequencing, samples were prepared from separate flasks (each containing 5×106 cells and infected with the same virus stock). The Sequel and the various MinION libraries have been prepared using the same RNA mixture.
Ribosomal RNA depletion For the Illumina sequencing and for the PacBio random-primed sequencing, the total RNA samples were depleted from rRNA using the Epicentre Ribo-Zero™ Magnetic Kit H/M/R (Illumina).
Selection of PolyA(+) RNA For the PacBio and MinION polyA sequencing, the polyA(+) fraction of the RNA samples were isolated using the Qiagen Oligotex mRNA Mini Kit, following the “Spin Columns” protocol.
PolyA purified and rRNA depleted RNA samples were quantified through use of the Qubit RNA HS Assay Kit (Life Technologies) and then subjected to cDNA synthesis according to the downstream applications.
cDNA synthesis, library preparation and sequencing Illumina sequencing
Total RNA was purified from PK-15 cells in various stages of PRV infection from 1 to 24 h pi and then, the samples were mixed together to uncover an extensive variety of viral transcripts. Libraries were prepared from ribo-depleted samples using the ScriptSeq v2 RNA-Seq Library Preparation Kit (Epicentre/Illumina) according to the manufacturer’s recommendations. The kit uses a random-primed (random hexamer with tagging sequence) cDNA synthesis reaction; this “original” protocol was used to construct a paired-end library, but for PolyA sequencing (PA-seq), a single-end library was prepared through the use of custom anchored adaptor-primer oligonucleotides with an oligo(VN)T20 primer sequence (Table 5). Briefly, the rRNA-depleted RNA samples were mixed with the primer (random or oligo(d)T) and the RNA Fragmentation Solution (part of the ScriptSeq Kit), and the mixtures were incubated at 85 °C for 5 min. The following kit components were mixed together: cDNA Synthesis Premix, DTT and StarScript AMV Reverse Transcriptase. This reagent mix was added to the pre-heated RNA-mixtures and they were incubated at 25 °C for 5 min and then 42 °C for 20 min. Reactions were cooled down to 37 °C. Finishing solution was added to the samples and incubation was continued at 37 °C for an additional 10 min, then at 95 °C for 3 min. Samples were cooled down to 25 °C, and Terminal Tagging Premix, as well as DNA Polymerase were added. The incubation was continued at 25 °C for 15 min and then at 95 °C for 3 min. The di-tagged cDNA samples were purified by using the AMPure XP beads (Beckman Coulter). The purified samples were amplified by PCR using the FailSafe PCR Premix E, primers and FailSafe PCR enzyme (Lucigen, Epicentre). The PCR conditions were as follows: initial denaturation at 95 °C for 1 min, followed by 15 cycles at 95 °C for 30 sec 55 °C for 30 sec and 68 °C for 3 min. The final incubation step was carried out at 68 °C for 7 min. The PCR amplicons were purified with AMPure beads. The quantity and the quality of the samples were checked using Qubit fluorometer (Life Technologies) and Agilent 2100 Bioanalyzer, respectively.
PacBio SMRTbell library preparation & sequencing – the non-amplified method & RSII sequencing
Generation of cDNAs
The SuperScript Double-Stranded cDNA Synthesis Kit (Life Technologies) was used to prepare cDNAs from the polyA(+) RNA samples. These samples were used to quantify the PRV transcriptome during the infection period between 1-12 h. The enzyme-which was included in the kit-was changed to SuperScript III Reverse Transcriptase. The first-strand synthesis reactions were primed with Anchored Oligo(dT)20 primers (Life Technologies, Table 5). The obtained cDNAs were measured by using Qubit HS dsDNA Assay Kit (Life Technologies). The total amount of cDNA synthesized at each time point was used to prepare SMRTbell templates.
Preparation of SMRTbell libraries-“Barcoding method”
The cDNA samples (~500 ng/sample) were used to prepare SMRTbell templates by using the PacBio DNA Template Prep Kit 1.0 following the Pacific Biosciences’ 2 kb Template Preparation and Sequencing protocol.
Repairing the cDNA ends
Template Prep Buffer, ATP High, dNTP and End Repair Mix (PacBio) were added to the samples and then they were incubated at 25 °C for 15 min.
Sample purification 0.6x volume
AMPure PB bead was added to the samples. They were mixed using VWR vortex mixer for 10 min at 2000 rpm and room temperature. Tubes were placed in a magnetic bead rack for 3 min. After the bead pellets were formed, the supernatant were discarded. Beads were 2-times washed with freshly prepared ethanol (70%). Samples were dried and then they were eluted in 30 μl Elution Buffer (PacBio).
Adapter ligation
This step was carried out at 25 °C for 15 min with the addition of specific bar-coded adapters (Table 6), Template Prep Buffer, ATP Low and Ligase (PacBio). The enzyme was inactivated at 65 °C (10 min).
Exonuclease treatment
ExoIII (50U) and ExoVII (5U) enzymes were added to the carrier DNA-cDNA mixture, then they were incubated at 37 °C for 1 h, then the reactions were returned to 4 °C.
Sample purification
SMRTbell Templates were purified using 0.6× AMPure PB beads, as was described above. Two purification steps were applied after one other. The final elution volume was 10 μl. Qubit fluorometer was used for quantitation.
SMRTbell templates were bound to the PacBio’s P5 DNA polymerase. These complexes were bound to MagBeads using the Pacific Biosciences MagBead Binding Kit. The concentrations of the SMRTbell libraries were measured by Qubit and they were also qualified by Agilent 2100 Bioanalyzer. The PacBio RSII platform and C3 sequencing chemistry was used for sequencing. 180 min movies were applied for each SMRT Cell.
Annealing of the sequencing primers to the template DNA and the DNA polymerase binding
The PacBio Calculator v.2.3.0.0. was used to set the annealing and binding reactions. 2000 bp insert size and 1 ng/μl concentration was set. The Sequencing Primer (5000 nM) was diluted to 150 nM with the PacBio Elution Buffer (EB). One μl from the diluted primer and 10x Primer Buffer were added to the template DNA. Annealing was carried out at 80 °C for 2 min, and then the temperature was ramp to 25 °C at a rate of 0.1 °C/sec. The total volume of annealed template was bound to the Polymerase. For this, 2 μl dNTP, 2 μl DTT, 2 μl Binding Buffer (BB) and 2 μl diluted Polymerase were added to the samples. Mixtures were incubated at 30 °C for 4 h and then they were heated to 37 °C for 30 min. 2 μl from the complexes were used for MagBead binding. Cleaned MagBeads (74 μl) were added to the samples and they were incubated at 4 °C for 2 h on a HulaMixer rotator (Invitrogen). After the incubation, samples were washed with 19 μl BB, then 19 μl Wash Buffer (WB), and finally they were resuspended in 19 μl BB. The total amount of the MagBead-bound complex was loaded onto the RSII sequencer.
Preparation of SMRTbell libraries-“Carrier DNA method”
The total amount of cDNA synthesized at each time point was used to prepare SMRTbell templates by using the PacBio DNA Template Prep Kit 1.0 and following the Pacific Biosciences template preparation and sequencing. protocol for Very Low (10 ng) Input 2 kb libraries with carrier DNA (pBR322, Thermo Scientific).
Preparing the carrier DNA
The concentration of the pBR322 plasmid DNA was measured by Nanodrop. A 100ng/μl stock solution was prepared from the plasmid using the PacBio Elution Buffer (EB). The DNA was exonuclease treated with the PacBio ExoIII (200U) and ExoVII (20U) enzymes and the Template Prep buffer (10×). The mixture was incubated at 37 °C for 1 h, then it was cooled down to 4 °C. The DNA was purified and concentrated by using 0.6× AMPure® PB beads and it was eluted in 50 μL EB. The exonuclease-treated carrier DNA was quantified by Qubit fluorometer.
Repairing the DNA damage
cDNA samples were mixed with DNA Damage Repair Buffer, NAD+, ATP High, dNTP and DNA Damage Repair Mix (all from the PacBio DNA Template Prep Kit), and then were incubated at 37 °C for 20 min. Samples were cooled to 4 °C.
Repairing the cDNA ends
End Repair Mix (PacBio) was added to the samples and then they were incubated at 25 °C for 5 min.
Sample purification 0.6x volume
AMPure PB bead was added to the samples and they were purified as in case of the barcoded samples.
Adapter ligation
This step was carried out at 25 °C for 60 min with the addition of Blunt Adapter, Template Prep Buffer, ATP Low and Ligase (PacBio). The enzyme was inactivated at 65 °C (10 min).
After this step, the ExoIII and ExoVII-treated carrier DNA (5 μl; 100 ng/μl) was mixed with the adapter-ligated cDNA samples (40 μl).
Exonuclease treatment
ExoIII (50U) and ExoVII (5U) enzymes were added to the carrier DNA-cDNA mixture, then they were incubated at 37 °C for 1 hour, then the reactions were returned to 4 °C.
Sample purification
Two purification steps were carried out successively, as was described earlier.
SMRTbell libraries were bound to DNA polymerases by using the DNA polymerase binding kit P5 and v2 sequencing primers. The DNA polymerase/template complexes were bound to MagBeads using the MagBead Binding Kit. The concentrations of the SMRTbell libraries were measured by Qubit and they were further analysed by Agilent 2100 Bioanalyzer. The cDNA sequencing reactions were carried out on the PacBio RSII platform with C3 sequencing chemistry with 180 min movies.
Annealing of the sequencing primers to the template DNA and the DNA polymerase binding
Conditioning and annealing of the Sequencing Primer, the binding of the Polymerase to the libraries, as well as Polymerase-template complex binding to the magnetic beads was done exactly as indicated by the PacBio Very Low Input protocol. The total amounts of prepared libraries (10 μl) were used for the binding. The DNA concentrations were set to 0.1 μl in the Calculator version 2.3.0.0. The “small-scale” preparation protocol and the “non-standard” protocol were chosen. The Sequencing Primer (5000 nM) was diluted to 150 nM in EB. One μl from the diluted primer and 10x Primer Buffer were added to the template DNA. Annealing was carried out at 80 °C for 2 min then the temperature was ramp to 25 °C at a rate of 0.1 °C/sec. The total volume of annealed template was bound to the Polymerase. For this, 2 μl dNTP, 2 μl DTT, 2 μl BB and 2 μl diluted Polymerase were added to the samples. Mixtures were incubated at 30 °C for 4 h and then they were heated to 37 °C for 30 min. The total volume from the polymerase binding step was used for MagBead binding. The salt molarity was adjusted for optimal binding by adding WB (0.3× volume) to the bound complex instead of BB. Cleaned MagBeads (26 μl) were added to the samples and they were incubated at 4 °C for 30 min on a HulaMixer rotator (Invitrogen). After the incubation, samples were washed with 26 μl BB, then 26 μl BW, and finally they were resuspended in 19 μl BB. The total amount of the MagBead-bound complex was loaded onto the PacBio machine.
PacBio SMRTbell library preparation-Iso-Seq method/the amplified protocol & sequencing on RSII as well as Sequel platforms
Full-length cDNAs were generated using the Clontech SMARTer PCR cDNA Synthesis Kit based on the PacBio Isoform Sequencing (Iso-Seq) protocol. No Size Selection method was carried out for the analysis of short viral transcripts, while Manual Agarose-gel Size Selection, as well as SageELF™ and BluePippin™ Size-Selection Systems (Sage Science) were used for the isolation of long RNA molecules. The first-strand cDNAs were generated by using the SMARTer PCR cDNA Synthesis Kit (Clontech), the reactions were primed with oligo(dT) (part of the Clontech Kit) or adapter-linked GC-rich random primers (ordered from IDT DNA). The single-stranded cDNAs were PCR-amplified using KAPA HiFi Enzyme (Kapa Biosystems), in accordance with recommendations provided by PacBio, as follows: initial denaturation was carried at −95 °C for 2 min, followed by 16 cycles for PA-seq, 20 or 30 cycles for random-primed samples (the optimal cycle was determined in the optimization step) at −98 °C for 20 s (denaturation), −65 °C for 15 s (annealing) −72 °C for 4 min (extension). The final extension was carried out at −72 °C for 5 min. (n: 16 cycles was ideal for the No size-selection protocol. For the agarose size-selection, 12 cycles and 1:45 min extension was set for the amplification of transcripts between 2–3 kb and 15 cycles and 3 min extension was used for the longer transcripts. Sixteen cycles were set for the SageELF and BluePippin samples. PCR products were pooled then size selected manually by using 0.8% agarose gel or with the SageELF™ System according to the PacBio's protocol. Size-selected samples were amplified with KAPA enzyme using the conditions as above. The fraction of cDNAs with a size over 5 kb was run on BluePippin™ System to eliminate the short SMRTbell libraries. Five-hundred ng of each non-size-selected cDNA sample was applied for the SMRTbell template preparation, using the PacBio DNA Template Prep Kit 1.0. The amount of cDNAs from the size-selected samples used in the library preparation reaction were based on the following PacBio protocols: Procedure & Checklist – Isoform Sequencing (Iso-Seq™) using the Clontech SMARTer PCR cDNA Synthesis Kit and (a) Manual Agarose-gel Size Selection; (b) SageELF™ Size Selection System; and (c) BluePippin™ Size-Selection System. SMRTbell sequencing libraries were bound to polymerases by using the DNA/Polymerase Binding Kit P6 and v2 primers. The polymerase-template complexes were bound to MagBeads with the PacBio MagBead Binding Kit. The qualities of the samples were checked on the Agilent 2100 Bioanalyzer. Sequencing reactions were performed by using the PacBio RS II sequencer with DNA Sequencing Reagent 4.0. Movie lengths were 240 min or 360 min (one movie was recorded for each SMRT Cell).
The volume of the sequencing primer for the annealing, and the polymerase (P5 or P6) for the binding was determined using the PacBio Calculator version 2.3.1.1., by adding the concentrations and the average insert sizes of SMRTbell templates.
The polymerase-template complexes were bound to MagBeads, loaded onto SMRT Cells and sequenced on the RSII instrument.
The PacBio’s Binding Calculator was used to prepare the library for sequencing using the MagBead one-cell per well (OCPW) protocol, and binding kit P6v2 was used with an on-plate concentration of 0.05 nM. The insert sizes were set according to the size-selections which were applied: 1000, 2500 and 6000 bp sizes were chosen.
In short, the sequencing primer was diluted in PacBio EB to 150 nM. The annealing step was performed with 1 μl template DNA (cc: ~20 ng/μl), the diluted sequencing primer and primer buffer (10x). The final concentration of this mixture was 0.8333 nM. Annealing was carried out at 20 °C for 30 min then the DNA polymerase enzyme was diluted to a final concentration of 50 nM in PacBio BB v2, and then it was bound to the annealed template followed by the addition of DTT, dNTP and BB. The complex (0.5 nM final concentration) was incubated at 30 °C for 4 h. The sample complex (0.5 μl) was mixed with and 18.5 μl MagBead Binding Buffer (0.0125 nM final concentration). MagBeads were prepared as follows: 73.9 μl MagBeads were washed with 73.9 μl MagBead WB, then 73.9 μl MagBead BB was added. The sample complex was bound to the washed, prepared MagBeads for loading to the RSII machine: 19 μl sample complex was added to the beads, and then it was placed at 4 °C for 30 min in a HulaMixer. After incubation, the MagBead-bound complex was washed with 19 μl BB, then with 19 μl WB and finally, it was resuspended in 19 μl BB. The total amount of the MagBead-bound complex was loaded onto the instrument. The MagBead One Cell Per Well protocol was used. One SMRT Cell was also run on Sequel instrument.
Oxford Nanopore cDNA sequencing
PRV transcripts were sequenced on MinION device using the 1D Strand switching cDNA by ligation method (Version: SSE_9011_v108_revS_18Oct2016) and the ONT Ligation Sequencing Kit 1D (SQK-LSK108). For this, PolyA(+)-selected RNAs were used. 50ng from the samples were subjected to reverse transcription. Poly(T)-containing anchored primer [(VN)T20; ordered from Bio Basic, Canada, (Table 5)] and dNTPs (10 mM, Thermo Scientific) was added to the RNA samples and then the mixture was incubated at 65 °C for 5 min. Buffer and DTT from SuperScipt IV Reverse Transcriptase kit (Life Technologies), RNase OUT (Life Technologies) and strand-switching oligo with three O-methyl-guanine RNA bases (PCR_Sw_mod_3G; ordered from Bio Basic, Canada) were added and the sample was incubated at 42 °C for 2 min. 200U SuperScript IV Reverse Transcriptase enzyme was measured into the mix. First-strand cDNA synthesis was carried out at 50 °C for 10 min; it was followed by the strand switching step at 42 °C for 10 min. Enzymes were inactivated at 80 °C for 10 min. Five μl from the prepared double-stranded cDNA was amplified in a single PCR reaction using KAPA HiFi DNA Polymerase (Kapa Biosystems) and Ligation Sequencing Kit Primer Mix (provided by the 1D Kit). The Veriti Thermal Cycler (Applied Biosystems) was set as the 1D Kit’s protocol recommended: initial denaturation for 30 sec at 95 °C (1 cycle); denaturation for 15 sec at 95 °C (15 cycles); annealing for 15 sec at 62 °C (15 cycles); elongation for 4 min at 65 °C (15cycles); final extension 10 min at 65 °C. NEBNext End repair / dA-tailing Module (New England Biolabs) was used for end repair, while NEB Blunt/TA Ligase Master Mix (New England Biolabs) was applied for adapter ligations. The adapter sequences were supplied by the kit. Agencourt AMPure XP magnetic beads (Beckman Coulter) were used for purification following each enzymatic step. The Qubit Fluorometer (Life Technologies Qubit 2.0) and the Qubit (ds)DNA HS Assay Kit were used to quantify the concentration of the libraries. Samples were loaded on R9.4 SpotON Flow Cells, and base calling was performed using Albacore v1.2.6.
Oxford Nanopore sequencing on Cap-selected samples
To obtain full-length transcripts with the exact 5′-ends, Cap selection was carried out. For this, the TeloPrime Full-Length cDNA Amplification Kit (Lexogen) was used, which has an exceptional specificity for 5′-Cap. The starting material was 2 μg total RNA diluted in 12 μl water, from a mixed PRV sample (containing RNA from 1, 2, 3, 4, 5, 6, 7, 8, 12, 18 and 24 h post-infection). The method based on cDNA generation. Reverse transcription (RT) was carried out according to the kit’s manual. Briefly, the diluted RNA was mixed with RT buffer, primer (both are supplied by the kit). The RT primer contains an “oligodT” sequence (Table 5) to select the polyadenylated transcripts. The mixture was preheated at 70 °C for 30 sec, then it was cooled down to 37 °C for 1 min. RT enzyme and reagents (part of the kit) were added and the reaction was contain at 37 °C for 2 min. Temperature was increased to 46 °C for 50 min. The RNA-cDNA hybrid was purified using silica columns (kit’s component). A specific adapter was ligated to the cDNA by base-pairing of the 5’C to the cap structure of the RNA. This step was carried out by the double-strand specific ligase of the kit. Ligation was performed at 25 °C, overnight. The sample was purified after ligation using the silica columns. The cDNA was converted to dscDNA using the Second-Strand Mix and the Enzyme Mix from the Teloprime kit. The reaction was carried out in a Veriti Cycler with the following protocol: 98 °C for 90 sec, 62 °C for 60 sec, 72 °C for 5 min, hold at 25 °C.
Sample concentration was measured using Qubit dsDNA HS Assay Kit (Life Technologies). Specificity of the obtained cDNA was checked by qPCR (Rotor-Gene Q) using a gene specific primer (us9, 10μM each; Table 7), cDNA and ABsolute qPCR SYBR Green Mix (Thermo Fisher Scientific) in 20 μl final volume. The initial denaturation was 94 °C 15 min, and it was followed by 35 cycles of 94 °C for 25 sec, 60 °C 25 sec and 72 °C 6 sec.
The PolyA(+)-CAP-selected samples were also sequenced on MinION using the 1D Strand switching cDNA by ligation method. These samples were subjected to the end repair and adapter ligation steps, and then they were loaded on the ONT Flow Cells.
Oxford Nanopore direct RNA sequencing
Three flow cells were used for sequencing PRV samples following the Direct RNA sequencing (DRS) protocol from the ONT (Version: DRS_9026_v1_revM_15Dec2016). Total RNAs from 12 different time points were mixed together, and then polyA selection was carried out. RNA from the PolyA(+) fraction in 9 μl was used as a template for sequencing. RNA was mixed with the RT (oligodT-containing T10) adapter (supplied by the ONT Direct RNA Sequencing Kit; SQK-RNA001; Oxford Nanopore Technologies) and T4 DNA ligase (2M U/ml; New England BioLabs). The mixture was incubated at room temperature for 10 min. First-strand cDNA synthesis was carried out in 40 μl final volume with SuperScript III Reverse Transcriptase (Life Technologies), according to the DRS protocol, at 50 °C for 50 min, then 70 °C for 10 min in a Veriti Thermal Cycler. Samples were washed with Agencourt AMPure XP Beads (Beckman Coulter). XP Beads were treated before usage with RNase OUT (40 U/μl; Life Technologies); 2U enzyme was added to 1 μl bead. Purified RNA-cDNA hybrids were eluted in 20 μl Ambion Nuclease-Free Water (Thermo Fisher Scientific). RMX sequencing adapter was ligated to the eluted samples with T4 DNA ligase and NEBNext Quick Ligation Reaction Buffer (New England BiceoLabs) at room temperature for 10 min. Samples were purified with RNase OUT-treated XP beads using Wash Buffer (part of the DRS Kit) and then eluted in 21 μl Elution Buffer (provided by the DRS Kit). The concentration of the reverse-transcribed and adapted RNA was measured by using the Qubit 2.0 Fluorometer and Qubit dsDNA HS Assay Kit (Life Technologies). Samples were loaded onto the R9.4 SpotON Flow Cell.
Data on the quality of PacBio RSII, Sequel, and ONT MinION reads including insertions, deletions, and mismatches, as well as the coverages are summarized in Table 8 (available online only).
Read processing
Raw reads from the random-primed Illumina sequencing were aligned to the PRV genome (KJ717942.1), using Tophat v2.09 (ref. 23); ambiguous reads were discarded. For PA-Seq, mapping was carried out with Bowtie v2 (ref. 24).
The PacBio RSII and Sequel consensus reads were generated following the RS_ReadsOfInsert protocol of the SMRT Analysis (v2.3.0 and v5.0.0) (Fig. 2), with the following settings: Minimum Full Passes=1, Minimum Predicted Accuracy=90, Minimum Length of Reads of Insert=1, Maximum Length of Reads of Insert=No Limit. These consensus reads were mapped using GMAP25, with the following settings: gmap -d Genome.fa --nofails -f samse File.fastq>Mapped_file.sam.
The ONT's Albacore software (v.2. 0.1) was used for base calling. This basecaller identify the nucleotide sequences directly from raw data. The sequencing reads were mapped with GMAP using the same setting as was described above.
Custom routines were used to acquire the quality information presented in this data descriptor. The codes have been archived on Github (https://doi.org/10.5281/zenodo.1034511).
Data Records
All sequencing data have been uploaded to the European Nucleotide Archive under the project accession PRJEB24593 (Data Citation 1)-contains BAM files-and PRJEB9526 (Data Citation 2) – containing FASTQ files -. All sequencing reads were mapped to the KJ717942.1 genome build. All data can be used without restrictions.
Technical Validation
The quantity of the isolated total RNAs, the polyA-selected RNAs, the rRNA-depleted samples, as well as the synthesized cDNA fractions and sequencing-ready libraries were measured by Qubit 2.0 (Life Technologies) fluorometer using the Qubit RNA, HS RNA and HS dsDNA Assay Kits. The conditions for primer annealing and binding of the polymerase to the templates were determined by PacBio’s Binding Calculator in RS Remote. The libraries were measured by Agilent 2100 Bioanalyzer using the Agilent High Sensitivity DNA Kit.
Usage Notes
The provided dataset was primarily produced to discover and determine the complexity and expression dynamic properties of PRV transcriptome. The uploaded binary alignment (BAM) files contain reads already mapped to the KJ717942.1 reference. These aligned files can be further analysed using various bioinformatics program packages, such as bedtools26, samtools27, or visualized using e.g. IGV28, Geneious29 or Artemis30. The uploaded Illumina, PacBio and ONT files have not been trimmed, they contain terminal poly(A) sequences as well as the 5′and 3′ adapter sequences, which can be used to determine the orientations of the reads.
Additional information
How to cite this article: Tombácz, D. et al. Transcriptome-wide survey of pseudorabies virus using next- and third-generation sequencing platforms. Sci. Data 5:180119 doi: 10.1038/sdata.2018.119 (2018).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
References
Aujeszky, A. A contagious disease, not readily distinguishable from rabies, with unknown origin. Veterinarius 12, 387–396 (1902).
Pomeranz, L., Reynolds, A. & Hengartner, C. Molecular biology of pseudorabies virus: Impact on neurovirology and veterinary medicine. Microbiol Mol Biol Rev. 69, 462 (2005).
Boldogkői, Z. & Nógrádi, A. Gene and cancer therapy--pseudorabies virus: a novel research and therapeutic tool? Curr Gene Ther. 3, 155–182 (2003).
Yang, M. et al. Retrograde, transneuronal spread of pseudorabies virus in defined neuronal circuitry of the rat brain is facilitated by gE mutations that reduce virulence. J. Virol. 73, 4350–4359 (1999).
Song, C. K., Enquist, L. W. & Bartness, T. J. New developments in tracing neural circuits with herpesviruses. Virus Res. 111, 235–249 (2005).
Boldogkői, Z. et al. Genetically timed, activity-sensor and rainbow transsynaptic viral tools. Nat. Methods 6, 127–130 (2009).
Granstedt, A. E., Kuhn, B., Wang, S. S. & Enquist, L. W. Calcium imaging of neuronal circuits in vivo using a circuit-tracing pseudorabies virus. Cold Spring. Harb. Protoc 2010, pdb.prot5410 (2010).
Card, J. P. et al. A dual infection pseudorabies virus conditional reporter approach to identify projections to collateralized neurons in complex neural circuits. PLoS ONE 6, e21141 (2011).
Zhu, L. et al. Growth, physicochemical properties, and morphogenesis of Chinese wild-type PRV Fa and its gene-deleted mutant strain PRV SA215. Virol. J. 8, 272 (2011).
Maresch, C. et al. Oral immunization of wild boar and domestic pigs with attenuated live vaccine protects against pseudorabies virus infection. Vet. Microbiol. 161, 20–25 (2012).
Klingbeil, K. et al. Immunization of pigs with an attenuated pseudorabies virus recombinant expressing the hemagglutinin of pandemic swine origin H1N1 influenza A virus. J. Gen. Virol. 95, 948–959 (2014).
Oláh, P. et al. Characterization of pseudorabies virus transcriptome by Illumina sequencing. BMC Microbiol. 15, 130 (2015).
Tombácz, D. et al. Strain Kaplan of Pseudorabies Virus Genome Sequenced by PacBio Single-Molecule Real-Time Sequencing Technology. Genome Announc 2, 14–15 (2014).
Rhoads, A. & Fai, K. PacBio Sequencing and Its Applications. Genomics, Proteomics &. Bioinformatics 13, 278–289 (2015).
Sharon, D., Tilgner, H., Grubert, F. & Snyder, M. A single-molecule long-read survey of the human transcriptome. Nat. Biotechnol. 31, 1009–1014 (2013).
Tilgner, H., Grubert, F., Sharon, D. & Snyder, M. P. Defining a personal, allele-specific, and single-molecule long-read transcriptome. Proc. Natl. Acad. Sci. USA 111, 9869–9874 (2014).
Chen, S. Y., Deng, F., Jia, X., Li, C. & Lai, S. J. A transcriptome atlas of rabbit revealed by PacBio single-molecule long-read sequencing. Sci. Rep 7, 7648 (2017).
Tombácz, D. et al. Full-Length Isoform Sequencing Reveals Novel Transcripts and Substantial Transcriptional Overlaps in a Herpesvirus. PLoS ONE 11, e0162868 (2016).
Moldován, N. et al. Multi-Platform Sequencing Approach Reveals a Novel Transcriptome Profile in Pseudorabies Virus. Front. Microbiol 8, 2708 (2018).
Tombácz, D. et al. Characterization of the Dynamic Transcriptome of a Herpesvirus with Long-read Single Molecule Real-Time Sequencing. Sci. Rep 7, 43751 (2017).
Lomniczi, B., Watanabe, S., Ben-Porat, T. & Kaplan, A. S. Genetic basis of the neurovirulence of pseudorabies virus. J. Virol. 52, 198–205 (1984).
Kaplan, A. S. & Vatter, A. E. A comparison of herpes simplex and pseudorabies viruses. Virology 7, 394–407 (1959).
Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009).
Langmead, B. & Salzberg, S. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 9, 357–359 (2012).
Wu, T. D. & Watanabe, C. K. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21, 1859–1875 (2005).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011).
Kearse, M. et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012).
Rutherford, K. et al. Artemis: sequence visualization and annotation. Bioinformatic 16, 944–945 (2000).
Weirather, J. L. et al. Comprehensive comparison of Pacific Biosciences and Oxford Nanopore Technologies and their applications to transcriptome analysis. F1000Research 6, 100 (2017).
Schirmer, M., D’Amore, R., Ijaz, U. Z., Hall, N. & Quince, C. Illumina error profiles: resolving fine-scale variation in metagenomic sequencing data. BMC Bioinformatics. 17, 125 (2016).
Data Citations
European Nucleotide Archive PRJEB24593 (2018)
European Nucleotide Archive PRJEB9526 (2015)
Acknowledgements
This work was supported by the Swiss-Hungarian Cooperation Programme: SH/7/2/8 to ZBo. The work was also supported by the Bolyai János Scholarship of the Hungarian Academy of Sciences to DT and by the NIH Centers of Excellence in Genomic Science (CEGS) Center for Personal Dynamic Regulomes: 5P50HG00773502 to MS.
Author information
Authors and Affiliations
Contributions
D.T. performed the Illumina, PacBio and ONT sequencing, analyzed the data and drafted the manuscript. D.S. performed the PacBio sequencing. A.S. conducted bioinformatics analysis. N.M. took part in ONT sequencing experiments. M.S. conceived and designed the experiments. Z.B. conceived and designed the experiments, coordinated the project and wrote the final manuscript. All authors read and approved the final paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
ISA-Tab metadata
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files made available in this article.
About this article

Cite this article
Tombácz, D., Sharon, D., Szűcs, A. et al. Transcriptome-wide survey of pseudorabies virus using next- and third-generation sequencing platforms. Sci Data 5, 180119 (2018). https://doi.org/10.1038/sdata.2018.119
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/sdata.2018.119
This article is cited by
-
Identification of herpesvirus transcripts from genomic regions around the replication origins
Scientific Reports (2023)
-
Time-course transcriptome analysis of host cell response to poxvirus infection using a dual long-read sequencing approach
BMC Research Notes (2021)
-
Time course profiling of host cell response to herpesvirus infection using nanopore and synthetic long-read transcriptome sequencing
Scientific Reports (2021)
-
Time-course profiling of bovine alphaherpesvirus 1.1 transcriptome using multiplatform sequencing
Scientific Reports (2020)
-
Long-read assays shed new light on the transcriptome complexity of a viral pathogen
Scientific Reports (2020)
