
Abstract
Metastatic colorectal cancer is a leading cause of cancer death. However, current therapy options are limited to chemotherapy, with the addition of anti-EGFR antibodies for patients with RAS wild-type tumours. Novel drug targets, or drug combinations that induce a synergistic response, would be of great benefit to patients. The identification of genes that are essential for cell survival can be undertaken using functional genomics screens. Furthermore, performing such screens in the presence of a targeted agent would allow the identification of combinations that result in a synthetic lethal interaction. Here, we present a dataset containing the results of a large scale RNAi screen (815 genes) to detect essential genes as well as synergistic combinations with targeted therapeutic agents using a panel of 27 colorectal cancer cell lines. These data identify genes that are essential for colorectal cancer cell survival as well as synthetic lethal treatment combinations using novel computational approaches. Moreover, this dataset could be utilised in combination with genomic profiling to identify predictive biomarkers of response.
Design Type(s) | screening campaign • strain comparison design |
Measurement Type(s) | cell viability |
Technology Type(s) | RNAi screening |
Factor Type(s) | colorectal cancer cell line |
Sample Characteristic(s) | Homo sapiens • colorectal cancer cell line |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others


Background & Summary
Colorectal cancer is a leading cause of cancer mortality in the UK1,2, with >15,000 deaths each year. While early diagnosis leads to significantly higher survival, the prognosis for advanced stage disease remains poor. Therapeutic options for advanced colorectal cancer are currently limited to chemotherapy regimens (FOLFOX or FOLFIRI), and the anti-EGFR antibodies cetuximab or panitumumab3,4. While anti-EGFR therapy is beneficial in a subset of patients, tumours with activating KRAS, NRAS or BRAF mutations are intrinsically resistant, and exhibit constitutive activation of the MEK/ERK and/or PI3K/AKT signalling pathways3,5,6.
Despite the development of targeted agents over the last few decades, effective strategies to treat late-stage colorectal cancer have not emerged. While the concept of inhibiting a single signalling molecule remains attractive, in practice the inhibition of multiple targets often results in a more durable response. Furthermore, through the simultaneous inhibition of multiple signalling pathways, combination therapy may allow synthetic lethal interactions to be harnessed, and overcome intrinsic resistance to targeted agents7–10.
Several recent studies have successfully utilised high-throughput RNAi screening for profiling the essential gene landscape, and the identification of novel drug targets for cancer therapy11–14. Further, both chemical and genomic screens for synthetic lethal combinations have successfully identified targetable cancer-specific sensitivities8,15–19. However, it is important to note that in order to properly address the genomic heterogeneity of cancer, multiple cell lines are required. As is commonly seen, failure to do so results in a dataset that fails to capture the genetic diversity observed in the clinic, and identifies hits that are relevant only to the one cell line used.
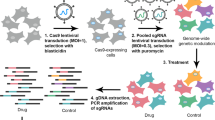
In this study, high-throughput functional genomics screens were used to identify and validate synthetic lethal combinations in a panel of colorectal cancer cell lines. An overview of the experimental design is shown in Fig. 1. The screen contained four experimental arms: the DMSO anchor arm, to assess the activity of siRNA alone, and the three ‘plus-drug’ arms where cells were also treated with one of three anchor drugs targeting EGFR (cetuximab), MEK (trametinib) or PI3K (BYL719). Twenty-seven colorectal cancer cell lines were transfected with a customised siRNA library targeting the human kinome as well as 95 genes commonly mutated in colorectal cancer (Data records 1 and 3). We elected to target kinases in our screen as they are potentially more amenable to drug development in the event of specific vulnerabilities being identified. Additionally, for some kinases chemical probes or indeed drugs may already exist. Following 72 h incubation with the siRNA libraries, cells were assessed for decreased ATP levels (using the CellTiter Glo assay (Promega)), as an indicator of decreased viability.

The primary kinome siRNA screen was performed using a panel of 27 colorectal cancer cell lines. A DMSO-treated arm allowed identification of ‘essential’ genes, while the ‘plus-drug’ arms enabled selection of synergistic combinations for validation in the secondary screen.
The primary siRNA screen hits were binned according to the screen arm: DMSO arm hits were binned as ‘Essential’ using a z-score cut-off of <−3, while ‘plus-drug’ arm hits were ranked according to their synergy score, and the number of cell lines that showed a synergistic response (Data records 1 and 3). Essential genes identified in the primary screen were enriched for genes connected to the cell cycle, and contained many known pan-cancer single gene vulnerabilities20–23 (Data records 1 and 3). A total of 37 genes were identified that scored <−3 across three or more different cell lines.
The top 38 drug/gene synergistic combinations were selected for confirmation in a secondary deconvolution screen (4 siRNA sequences per gene) (Data records 2 and 3). siRNAs targeting the ‘Essential’ genes PLK1 and SF3B1 were also included. Deconvoluted siRNAs targeting 40 genes were rescreened as previously against the DMSO arm and three ‘plus-drug’ arms (Supplementary Fig. 1). This secondary screen led to the validation of 8 drug/siRNA combinations, which reproduced the original synergistic phenotype with 2 or more individual siRNA in greater than 75% of the cell lines that showed synergy in the original primary screen (Data records 2 and 3). These strict validation criteria mean that only hits that were confirmed in multiple cell line models with multiple siRNAs are considered ‘validated’ with high confidence and should be prioritised in future investigations. As a result the datasets presented here will be of benefit to the fields of cancer biology, therapeutics, and molecular signalling, and further provides the broader high-throughput screening community with a robust method and screening dataset.
Methods
Cell lines
A panel of commercially available colorectal cancer cell lines24,25 were grown in either RPMI or DMEM/F12 medium, supplemented with 10% fetal calf serum and 1% penicillin/streptavidin, and maintained at 37 °C in a humidified atmosphere at 5% CO2. Cell lines used in these screens, and the number of cells seeded per well, are listed in Table 1. The number of cells per well was established as part of the Genomics of Drug Sensitivity in Cancer (GDSC) screening project. The dynamic range within the assay was maximised by determining the number of cells required to achieve the greatest intensity value, while also ensuring the growth of untreated cells was unrestricted by factors such as space and nutrients. A minimum of 6 seeding densities were tested under screening conditions using a two-fold dilution series. All cell lines are routinely SNP profiled to detect cross-contamination and STR profiled to confirm their identity with the providing repository.
Assay optimisation
Prior to screening we performed optimisation of assay conditions using a subset of 11 colorectal cancer cell lines. The type and concentration of transfection reagent was established by transfecting cells with Non-targeting siRNA pool #2 or si_PLK1 using six different transfection reagents (DharmaFECT 1, DharmaFECT 2, DharmaFECT 3, DharmaFECT 4 (all from Dharmacon), Lipofectamine RNAiMAX, and Lipofectamine 2,000 (both from Invitrogen)) at 4 different concentrations, with 4 different siRNA concentrations. The optimal transfection score was calculated for each cell line as follows:
This score takes into account any negative effect on cell viability that the transfection reagent may have, as well as rewarding conditions where the positive control siRNA (si_PLK1) is best at reducing cell viability compared to the control. The conditions with the best average OTS across all 11 lines were selected for screening.
The concentration of anchor drugs was selected by performing dose response curves for each drug against 16 colorectal cancer cell lines. As the aim was to identify synergistic drug/siRNA combinations, it was important to select an anchor drug concentration that had a minimal effect on cell viability alone. A dose of the cetuximab and BYL719 compounds was therefore selected that inhibited cell viability by less than 20% in more than 75% of cell lines. Colorectal cancer is often driven by activation of the MEK/ERK pathway, and many of the lines on which trametinib was tested showed sensitivity to MEK inhibition. The dose of trametinib was therefore selected to 1) demonstrate effective inhibition of MEK phosphorylation and 2) inhibit cell viability by <30% in more than 50% of the cell lines.
High throughput RNA interference screening
The protocol developed for this screen was adapted from previously published methods26, and is described below in detail. The primary siRNA screen was performed with biological replicates (see Supplementary Fig. 2 for number of replicates of each line), in 1,536-well plates. This enabled all 815 siRNA SMARTpools (4 siRNA pooled per gene) to be screened on one assay plate. A wide variety of positive and negative controls were located in specified wells across the plate, as depicted in Supplementary Fig. 1. The key positive controls for siRNA transfection were siRNA SMARTpools targeting PLK1 (GE Dharmacon, #M-003290-01); known to be important for cell cycle, and a propriety cell death siRNA control (siTOX) (GE Dharmacon, #D-001500-01). The broad kinase inhibitor staurosporine (2 uM) was also added to select wells of each plate as a positive control for treatment by the anchor compounds. Non-targeting siRNA pool #2 was utilised as a negative control for siRNA transfection (GE Dharmacon, #D-001206-14). Mock transfected (lipid only) wells were also added as a reference. Other controls that were included, but not used were siRNA SMARTpool targeting KIF11 (GE Dharmacon, #M-003317-01), Non-targeting siRNA sequence #2 (GE Dharmacon, #D-001210-02) and siGENOME RISC-Free Control siRNA (GE Dharmacon, #D-001220-01).
The custom SMARTpool siRNA library was designed to target 95 genes commonly mutated in colorectal cancer (Data Record 3). Of these 95 genes, 15 were already present in the kinome library, resulting in 15 duplicates, and overall 794 unique siRNAs (the kinome library also has 6 duplicates). Note that duplicate wells are not able to be reported in Pubchem (Data Record 1), but are included in the raw data files (Data Record 3).
The siRNA transfer and addition of each anchor drug were performed using an Echo 555 acoustic dispenser (Labcyte Inc.). An XRD-384 (FluidX) automated reagent dispenser was used for all other liquid handling steps.
Specialised consumable reagents that were required to perform the siRNA screens:
-
Primary screen: Dharmacon Human siGENOME SMARTpool siRNA Library—Protein Kinases (#G-003505-01, Lot 10,169), and a Dharmacon Human siGENOME Custom SMARTpool siRNA Library (see Data Record 3)
-
Secondary screen: Dharmacon Human siGENOME Custom siRNA Library—Set of 4 siRNA duplexes per gene
-
1,536-well plates, polystyrene, tissue-culture treated, clear flat bottom wells, sterile, with lid, black (Corning Costar, Tewksbury, MA, #3893)
-
Lipofectamine RNAiMAX Transfection Reagent (Invitrogen, #13778500)
-
OPTI-MEM (Gibco, #31985062)
-
CellTiter-Glo Luminescent Cell Viability Assay (Promega, #G7572)
Day 1: siRNA reverse transfection
siRNA library and control plates were thawed at room temperature 1 h prior to use. Cells were incubated in TrypLE (Life Technologies) until detached, then washed in growth media, counted and diluted to the desired concentration in antibiotic-free DMEM/F12 or RPMI media (Table 1).
For the siRNA transfection, 70 nl of the siRNA library (2.5 μM stock) was dispensed into each well of 1,536-well plates (Corning, #3893) (using an Echo 555 acoustic dispenser). Lipofectamine RNAiMAX transfection reagent was diluted in OPTI-MEM media (1:50) and incubated for 5 min, before dispensing 1.5 μl into each well of each plate.
Plates were then incubated for 20 min at room temperature, before dispensing 6 μl cells to each well (FluidX XRD-384 multiwell dispenser) (total well volume 7.5 μl). The final siRNA concentration was 23.9 nM.
Column 1 of each plate received media only (no cells) for background luminescence readings. The plate layout was designed so that edge wells were not used, and control wells (positive and negative) were spread across the plate (Supplementary Fig. 1).
Plates were incubated at 37 °C in a humidified atmosphere at 5% CO2 in a Cytomat 24C rotating incubator (Thermo Fisher Scientific) to minimise temperature gradients.
Day 2: Drug treatment
After 24 h incubation, 7.5 nl of anchor drug or vehicle (DMSO) alone was added to each well as appropriate. Cells were treated to achieve a final concentration of 5 μg ml−1 (32.9 μM) cetuximab (obtained from the Addenbrookes’ Hospital pharmacy), 10 nM trametinib (Selleckchem), or 1 μM BYL719 (Selleckchem). Vehicle treated wells received an equivalent volume of DMSO.
Day 4: Viability assay
To measure cellular ATP levels, the CellTiterGlo assay (Promega) was used. 2.5 μl of CellTiterGlo reagent was added to each well and incubated for 10 min. Luminescence was then measured using a Molecular Devices Paradigm plate reader. Changes in ATP levels were used as an indicator of overall cell viability.
Data analysis
The analysis of screen data entailed background correction, normalisation and scoring steps. Analysis was performed using custom R scripts, and is detailed below. Preliminary analysis of the raw data (Data Record 3) uncovered a relatively consistent diagonal viability gradient across each plate. The raw luminescence intensity readings for each plate of the primary screen were corrected for these position bias effects using a loess normalization approach27,28. This method was chosen as it performed better than the B-score method29, which we found to overfit the data and significantly increased the kurtosis of the dataset. Note that no correction method was required for the secondary screen data.
For each plate the luminescence intensity readings were background corrected by subtracting the mean value of blank wells. This removes any background noise that may result from the cell medium. Each well was then normalized to the mean of DMSO treated Non-targeting siRNA pool #2 negative control wells (24 wells) on that plate to obtain a relative viability score (Data records 1 and 3). Note that viability values were capped at a maximum of 1 in order to obtain meaningful Bliss additivity score values (below).
For quality control purposes, we calculated two Strictly Standardized Mean Difference (SSMD)30–32 values for each plate, using the two positive siRNA controls (siPLK1 and siTOX), and passed plates if either SSMD value was greater than 330. Using plates that passed the SSMD threshold, the biological replicate plates for each cell line were then averaged.
In order to identify hits in the DMSO anchor arm (i.e., effect of siRNA alone) z-scores were calculated on a per-cell line basis29. Z-score normalisation is used to scale the results to a standard normal distribution, using the mean and standard deviation of the experimental wells. This approach ensured that variation in siRNA transfection efficiency across the panel did not affect our ability to select important viability genes in each cell line.
To identify synergistic drug/siRNA combination hits, we calculated the Bliss additivity score33 for each drug/siRNA combination across each cell line as following (where V is the observed relative viability):
We then calculated the synergy score for each combination, by computing the difference between the expected Bliss additivity and the observed viability of the combination as following:
Each drug/siRNA combination was then ranked according to the number of cell lines where Synergy score > 0.15. We selected 38 top drug/siRNA combinations that showed strong synergy scores (Synergy score > 0.15), across three or more cell lines. We prioritised combinations that also resulted in lower overall viability. Many siRNAs that ranked highly with one anchor drug were also synergy hits with a second anchor, and so we designed the secondary screen so that all 38 siRNAs were rescreened against all three ‘plus-drug’ arms. The secondary screen utilised four siRNA sequences per gene, assayed separately (i.e., deconvoluted). The secondary screen data was then analysed using the same synergy score metric as previously, so that a synergy score was calculated for each individual siRNA duplex (Data records 2 and 3). For each siRNA duplex we determined whether the drug/siRNA combination reproduced a synergistic phenotype. The following threshold was used:
The number of siRNA duplex per cell line that scored as reproduced was then tallied for each combination.
Data Records
Data record 1
Primary siRNA screen data for all 27 cell lines are available at PubChem (Data Citation 1 to Data Citation 27). Assay ID accession numbers are provided in Table 2 (available online only). Screen-wide normalised data (negative control normalisation and z-score normalisation, where appropriate) are provided, as well as synergy scores for drug/siRNA combinations and the results of consequent binning strategies. The PubChem activity score indicates whether an siRNA was ‘active’ and binned as ‘Essential’ (designated 2, i.e., a screen hit in the DMSO arm) or ‘inactive’ (designated 1, i.e., not a screen hit). Samples are defined by siRNA catalogue number (Dharmacon) and Entrez Gene ID.
Data record 2
Secondary deconvolution siRNA screen data for 23 cell lines are available at PubChem (Data Citation 28 to Data Citation 50). Assay ID accession numbers are provided in Table 2 (available online only). Screen-wide normalised data (negative control normalisation) are provided, as well as synergy scores for drug/siRNA combinations and the results of consequent binning strategies. The PubChem activity score indicates whether an siRNA was ‘active’ and binned as ‘Synergy (designated 2, i.e., a screen hit in any ‘plus-drug’ arm) or ‘inactive’ (designated 1, i.e., not a screen hit). Samples are defined by siRNA catalogue number (Dharmacon) and Entrez Gene ID.
Data record 3
Raw data for both the Primary siRNA screen and the Secondary deconvolution screen are available at Figshare (Data Citation 51). Data for all assay plates are provided, including those with SSMD<3 that failed our QC threshold. Details of genes targeted by the custom siRNA library used in the Primary siRNA screen are also included, as are details of the siRNA/drug combinations selected for rescreening in the secondary deconvolution screen.
Technical Validation
Control performance and plate QC
The performance of positive and negative siRNA controls was quantified using the Strictly Standardised Mean Difference (SSMD)30–32 (Table 3); a statistical measure of the dynamic range between positive to negative controls, encompassing the mean and standard deviation of each control. Generally, a desirable SSMD for an RNAi screen is ≥3 in the context of a strong control30–32. We calculated two SSMD scores for each assay plate, using each of the positive siRNA controls (siTOX and siPLK1), compared with the negative control Non-targeting siRNA pool #2 (NTPool#2). While most cell lines responded equally well to both siTOX and siPLK1, there were some cell lines that did not respond to siTOX while siPLK1 had a large viability effect, and vice versa. Therefore, for each assay plate the higher of the two SSMD scores was used to pass or fail the plate. Plates that passed the quality control criteria had an average NTPool#2/siPLK1 SSMD of 4.23 and median of 4.09, and an average NTPool#2/siTOX SSMD of 4.12 and median of 4.16 (Table 3, Fig. 2) indicating very good data in the context of a strong positive control27.

Histogram of SSMD scores that passed the quality control threshold SSMD >3 in the (a) primary screen, and (b) secondary screen.
Assay plates for four cell lines consistently failed (Supplementary Fig. 2), and could be attributed to very low siRNA transfection efficiency, or, in the case of MDST8, a significant unexplained viability decrease in the presence of the Non-targeting siRNA controls. These cell lines were therefore excluded from the primary screen. Of the 416 total plates set up for the primary screen, a total of 310 passed our QC threshold (74.5%). In the secondary deconvolution screen 83 plates were assayed, of which 71 passed the QC threshold (85.5%). Two cell lines (HCC-56 and SNU-407) could not be rescreened in the secondary screen due to technical reasons.
Biological reproducibility across screening experiments
To assess the biological replicate plate reproducibility, the Pearson correlation coefficient was calculated for each set of duplicate plates per cell line (using all plates from DMSO and synergy arms). All cell lines were highly reproducible with an overall median correlation between biological replicates of 0.77 (standard deviation: 0.13) (Fig. 3).

Primary screen identified known candidates
The primary siRNA screen data (DMSO arm) returned a list of genes scored as essential (z-score<−3) for each cell line (Data record 1). Summarising this gene list showed that, as expected, PLK1 (polo like kinase 1) was identified as essential (z-score<−3) in 24 of the 27 cell lines, with a further 2 lines scoring<−2.9. Other genes that were scored as essential in many of the cell lines include the cell cycle kinases AURKA (aurora kinase A) and WEE1, and SF3B1 (mRNA splicing factor 3b subunit 1) (Fig. 4a).

(a) The distribution of siRNAs according to the average z-score across the panel of cell lines. Hits with low scores correspond to known cell cycle and essential genes. (b) Plot showing the z-scores of KRAS siRNA in cell lines with KRAS wild-type status versus KRAS mutant status. ***P value<0.001.
Cancer cells that harbour mutations in the KRAS gene are expected to be highly dependent on KRAS expression compared to KRAS wild-type lines. siRNA-mediated knockdown of KRAS was found to be lethal (z-score<−3) for 7 of 14 KRAS mutant lines compared to 1 of 13 KRAS wild-type cell lines (mean z-score KRAS mutant=−3.12, mean KRAS wild-type=−0.37; P=0.0002) (Fig. 4b). While we focused on identifying essential gene phenotypes that were observed across multiple cell lines, our dataset also allows the identification of vulnerabilities that are restricted to a single cell line. One example is the sensitivity of NCI-H716 to siRNA knockdown of FGFR2 (z-score=−4.31). Medico et al.34 recently reported that NCI-H716 has an amplification of FGFR2 and is sensitive to an FGFR inhibitor as a result.
The analysis of the primary screen (synergy arms) synergy scores showed that the combination of siRNA targeting PIK3CA with the anchor drug trametinib was identified as having a synergistic phenotype (Synergy score>0.15) in 7 cell lines (Data record 1), in keeping with the role of PI3K signalling in resistance to MEK1/2 (MAP2K1/2) inhibition35. While the reverse combination (BYL719 anchor drug with siRNA targeting MAP2K1) was not identified as synergistic, this is likely due to redundancy between MAP2K1 and MAP2K2. These results confirmed the ability of the screening methodology to identify known genes involved in colorectal cancer cell viability, and examples of synergistic drug/siRNA phenotypes.
Number of combinations validated in the deconvolution screen
The deconvolution screen was designed to primarily confirm the top synergistic combinations. We did however also deconvolute siRNAs targeting the top essential genes PLK1 and SF3B1. The results showed that individual siRNAs often produced stronger viability phenotypes than that observed using siRNA SMARTpools in the primary screen (Data record 2). In the Primary screen the effective concentration of each siRNA sequence in a SMARTpool is ~6 nM. However in the Secondary screen each individual siRNA sequence was 23.9 nM (4× higher concentration). While this may contribute to stronger viability phenotypes in the Secondary screen, part of the rationale for using siRNA SMARTpools is that by combining different siRNA sequences together the effective concentration of each siRNA sequence can be lower as the different siRNAs can act in concert.
The results of deconvoluted siRNAs validated the role of PLK1 (22/23 cell lines, where 2–4 of 4 siRNAs caused relative viability <0.5) and SF3B1 (16/23 cell lines, where 2–4 of 4 siRNAs caused relative viability <0.5) as essential genes in colorectal cancer. Of note is the fact that several genes (e.g., GUCY2D, CAMK2N1) that were ranked high in the primary screen (DMSO arm) list of essential genes were included in the secondary screen due to their role in synergistic combinations. Neither of these two genes were convincingly validated as single gene vulnerabilities (GUCY2D: 1/23 cell lines, and CAMK2N1: 1/23 cell lines) (Data record 2), despite a recent report which also identified these genes as essential genes in a pan-cancer siRNA screen11. This highlights the importance of a secondary deconvolution stage in any siRNA screening campaign.
The secondary screen validated a large proportion of synergistic drug/siRNA combinations. Table 4 summarises the top drug/gene combinations validating in the deconvolution screen. Over 75% of synthetic lethal combinations tested reproduced the primary screen synergy effect with at least two siRNA duplexes in at least one cell line. Fourteen combinations were highly validated with at least 2 siRNA duplexes, in three or more cell lines. Eight combinations were validated with at least 2 siRNA duplexes, in >75% of lines that showed the phenotype in the primary screen. This included siRNA targeting PIK3CA in combination with the anchor drug trametinib. Overall, the average synergy score for each combination across all cell lines in the secondary screen was well correlated with the average synergy score for that combination in the primary screen (Fig. 5). Mechanistic characterisation of novel synergistic combinations will be the subject of further publications.

Plots of average synergy scores of siRNA combinations with (a) BYL719, (b) cetuximab and (c) trametinib anchor.
Usage Notes
All siRNA screening data (Data records 1, 2 and 3) are provided so that users are able to investigate changes in viability and synergistic phenotypes by applying their own normalisation strategies and thresholds. This study focussed on identifying essential genes, across a heterogeneous panel of colorectal cancer cell lines. Genes that were scored as ‘Essential’ by siRNA knockdown alone (DMSO arm) can be investigated for their role in cell survival, with proof of principle being the lethality of PLK1 knockdown. The dependence of KRAS mutant cell lines on expression of KRAS, further demonstrates that the dataset will reveal associations between gene essentiality and genomic biomarkers.
In addition, we investigated and validated a number of novel synergistic drug/gene combinations using multiple siRNAs and multiple colorectal cancer cell lines. Only a subset of candidate combinations from the primary screen (synergy arms) were rescreened, and as the dataset contains the overall viability and synergy scores for each siRNA pool, further instances of synthetic lethal combinations may be uncovered. While the aim of this study was to identify drug/siRNA combinations that showed a synergistic response, investigators may also want to analyse the overall cell viability measurements in order to identify effective combinations that yield high cell death. In the clinic these combinations could also have potential benefit for patients.
Additional Information
How to cite this article: Williams, S. P. et al. High-throughput RNAi screen for essential genes and drug synergistic combinations in colorectal cancer. Sci. Data 4:170139 doi: 10.1038/sdata.2017.139 (2017).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
References
Ferlay, J. et al. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur. J. Cancer 49, 1374–1403 (2013).
Siegel, R., Desantis, C. & Jemal, A. Colorectal cancer statistics, 2014. CA Cancer J. Clin. 64, 104–117 (2014).
Pietrantonio, F. et al. First-line anti-EGFR monoclonal antibodies in panRAS wild-type metastatic colorectal cancer: A systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 96, 156–166 (2015).
Cremolini, C. et al. First-line chemotherapy for mCRC-a review and evidence-based algorithm. Nat. Rev. Clin. Oncol. 12, 607–619 (2015).
Walther, A. et al. Genetic prognostic and predictive markers in colorectal cancer. Nat Rev Cancer 9, 489–499 (2009).
De Roock, W., De Vriendt, V., Normanno, N., Ciardiello, F. & Tejpar, S. KRAS, BRAF, PIK3CA, and PTEN mutations: implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol. 12, 594–603 (2011).
Prahallad, A. et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 483, 100–103 (2012).
Vora, S. R. et al. CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell 26, 136–149 (2014).
Junttila, T. T. et al. Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell 15, 429–440 (2009).
Hatzivassiliou, G. et al. ERK inhibition overcomes acquired resistance to MEK inhibitors. Mol. Cancer Ther. 11, 1143–1154 (2012).
Campbell, J. et al. Large-Scale Profiling of Kinase Dependencies in Cancer Cell Lines. Cell Rep. 14, 2490–2501 (2016).
Davoli, T. et al. Functional genomics reveals that tumors with activating phosphoinositide 3-kinase mutations are dependent on accelerated protein turnover. Genes Dev. 30, 2684–2695 (2016).
He, T. et al. High-throughput RNAi screen in Ewing sarcoma cells identifies leucine rich repeats and WD repeat domain containing 1 (LRWD1) as a regulator of EWS-FLI1 driven cell viability. Gene 596, 137–146 (2017).
Siebring-van Olst, E. et al. A genome-wide siRNA screen for regulators of tumor suppressor p53 activity in human non-small lung cancer cells identifies components of the RNA splicing machinery as targets for anticancer treatment. Mol. Oncol. 11, 534–551 (2017).
Lee, J. et al. Comprehensive Two- and Three-Dimensional RNAi Screening Identifies PI3K Inhibition as a Complement to MEK Inhibitor AS703026 for Combination Treatment of Triple-Negative Breast Cancer. J. Cancer 6, 1306–1319 (2015).
Falkenberg, K. J. et al. A genome scale RNAi screen identifies GLI1 as a novel gene regulating vorinostat sensitivity. Cell Death Differ. 23, 1209–1218 (2016).
Falkenberg, K. J., Gould, C. M., Johnstone, R. W. & Simpson, K. J. Genome-wide functional genomic and transcriptomic analyses for genes regulating sensitivity to vorinostat. Sci. Data 1, 140017 (2014).
Jansen, V. M. et al. Kinome-wide RNA interference screen reveals a role for PDK1 in acquired resistance to CDK4/6 inhibition in ER-positive breast cancer. Cancer Res. 77, 2488–2499 (2017).
Manchado, E. et al. A combinatorial strategy for treating KRAS-mutant lung cancer. Nature 534, 647–651 (2016).
Blomen, V. A. et al. Gene essentiality and synthetic lethality in haploid human cells. Science 350, 1092–1096 (2015).
Wang, T. et al. Identification and characterization of essential genes in the human genome. Science 350, 1096–1101 (2015).
Hart, T. et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 163, 1515–1526 (2015).
Luo, B. et al. Highly parallel identification of essential genes in cancer cells. Proc. Natl. Acad. Sci. USA 105, 20380–20385 (2008).
Iorio, F. et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell 166, 740–754 (2016).
Garnett, M. J. et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 483, 570–575 (2012).
Chung, N. et al. A 1,536-well ultra-high-throughput siRNA screen to identify regulators of the Wnt/beta-catenin pathway. Assay Drug Dev. Technol. 8, 286–294 (2010).
Zhang, X. D . Optimal High Throughput Screening: Practical Experimental Design And Data Analysis For Genome Scale RNAi Research (Cambridge University Press, 2011).
Mpindi, J. P. et al. Impact of normalization methods on high-throughput screening data with high hit rates and drug testing with dose-response data. Bioinformatics 31, 3815–3821 (2015).
Birmingham, A. et al. Statistical methods for analysis of high-throughput RNA interference screens. Nat. Methods 6, 569–575 (2009).
Zhang, X. D. A pair of new statistical parameters for quality control in RNA interference high-throughput screening assays. Genomics 89, 552–561 (2007).
Zhang, X. D. et al. The use of strictly standardized mean difference for hit selection in primary RNA interference high-throughput screening experiments. J. Biomol. Screen. 12, 497–509 (2007).
Zhang, X. D. Novel analytic criteria and effective plate designs for quality control in genome-scale RNAi screens. J. Biomol. Screen. 13, 363–377 (2008).
Bliss, C. I. The toxicity of poisons applied jointly. Ann. Appl. Biol. 26, 585–615 (1939).
Medico, E. et al. The molecular landscape of colorectal cancer cell lines unveils clinically actionable kinase targets. Nat. Commun. 6, 7002 (2015).
Balmanno, K., Chell, S. D., Gillings, A. S., Hayat, S. & Cook, S. J. Intrinsic resistance to the MEK1/2 inhibitor AZD6244 (ARRY-142886) is associated with weak ERK1/2 signalling and/or strong PI3K signalling in colorectal cancer cell lines. Int. J. Cancer 125, 2332–2341 (2009).
Data Citations
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259276 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259299 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay /1259264 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259275 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259281 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259282 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259267 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259261 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259285 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259300 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259283 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259291 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259298 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259266 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259279 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259289 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259286 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259260 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259297 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259274 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259280 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259290 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259306 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259272 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259259 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259284 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259307 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259296 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259294 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259303 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259295 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259305 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259269 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259287 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259304 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259288 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259278 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259277 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259301 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259262 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259273 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259293 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259292 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259271 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259268 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259270 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259302 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259263 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259258 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. NCBI PubChem Bioassay 1259265 (2017)
Williams, S. P., Barthorpe, A. S., Lightfoot, H., Garnett, M. J., & McDermott, U. Figshare https://doi.org/10.6084/m9.figshare.5099959.v1 (2017)
Acknowledgements
We would like to thank members of the Garnett group for their technical assistance. We would also like to thank members of the McDermott, Garnett and Adams groups for helpful discussions and advice. The research leading to these results (S.P.W.) has received funding from the European Research Council under the European Union’s Seventh Framework Programme (FP7/2007-2013)/ERC synergy grant agreement n° 319661 COMBATCANCER. U.M. is supported by a Cancer Research UK Clinician Scientist Fellowship. M.J.G. is funded with an Award from the Wellcome Trust (102696).
Author information
Authors and Affiliations
Contributions
S.P.W. designed and performed experiments, conducted analyses, prepared figures and wrote the Data Descriptor. A.S.B. performed experiments and provided technical expertise. H.L. assisted with data processing. M.J.G. assisted in experimental design and provided intellectual input. U.M. provided project direction and intellectual support.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
ISA-Tab metadata
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files made available in this article.
About this article

Cite this article
Williams, S., Barthorpe, A., Lightfoot, H. et al. High-throughput RNAi screen for essential genes and drug synergistic combinations in colorectal cancer. Sci Data 4, 170139 (2017). https://doi.org/10.1038/sdata.2017.139
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/sdata.2017.139
