
Abstract
The Africanized honeybee (AHB) is a population of Apis mellifera found in the Americas. AHBs originated in 1956 in Rio Clara, Brazil where imported African A. m. scutellata escaped and hybridized with local populations of European A. mellifera. Africanized populations can now be found from Northern Argentina to the Southern United States. AHBs—often referred to as ‘Killer Bees’— are a major concern to the beekeeping industry as well as a model for the evolutionary genetics of colony defence. We performed high coverage pooled-resequencing of 360 diploid workers from 30 Brazilian AHB colonies using Illumina Hi-Seq (150 bp PE). This yielded a high density SNP data set with an average read depth at each site of 20.25 reads. With 3,606,720 SNPs and 155,336 SNPs within 11,365 genes, this data set is the largest genomic resource available for AHBs and will enable high-resolution studies of the population dynamics, evolution, and genetics of this successful biological invader, in addition to facilitating the development of SNP-based tools for identifying AHBs.
Design Type(s) | parallel group design • species comparison design |
Measurement Type(s) | whole genome sequencing |
Technology Type(s) | DNA sequencing |
Factor Type(s) | |
Sample Characteristic(s) | Apis mellifera • Municipality of Iaras |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others

Background & Summary
The European honeybee (Apis mellifera) was introduced to North and South America from Old World populations in the early 18th century1. In its native range, the honeybee is divided geographically and genetically into five ancestral lineages—the M and C lineages of Europe, the A lineage of Africa, and the Y and O lineages of Asia2–7—that encompass approximately 22 subspecies7.
European settlers, in the early 18th century, introduced subspecies of the M lineage (A. m. mellifera and A. m. iberica) into North America8,9. By the 20th century, C lineage (A. m. ligustica and A. m. carnica) and some O lineage (A. m. caucasia) subspecies were introduced8,9. It was during this century that Brazilian beekeepers first imported honeybees, chiefly A. m. mellifera and A. m. carnica, followed by A. m. ligustica and A. m. caucasia10. These subspecies were used exclusively in Brazil until 1956 when A. m. scutellata was imported from Africa for genetic research. Mated A. m. scutellata queens arrived from South Africa11 to breeding stations in Rio Claro, São Paulo, Brazil. The intention of the breeding program was to crossbreed A. m. scutellata with commercial stock to serve as a base population in selection programs12. Famously, queens and drones escaped and hybridized with the existing population12,13. These hybrids became one of the most astounding insect invaders in recent history; feral populations of the ‘Africanized’ bees retained the highly defensive trait of their African ancestors and are now the most common honeybee found from Central South America (Brazil and Northern Argentina) to Mexico and the southern United States14.
The sequencing of the honeybee genome in 2006 (ref. 15) was a landmark for the field of sociogenomics and has created valuable resources for the beekeeping industry16–19, but because this genome was derived from a typical admixed North American honeybee15 it provides little information about the underlying genetic variation present in Africanized populations.
Here, we present the genomes of 360 AHBs from Brazil along with a reference SNP database for this population. Genomic resources for AHBs will be beneficial for both pure and applied research questions. First, there is a growing need to quickly and reliably detect Africanized colonies to secure international trade in honeybees16,19. Second, Africanized bees are highly defensive and they are commonly used for studying the genetics of nest defence20. Finally, Africanized bees are highly invasive: within the last 60 years they have become the most common genotype across much of the Southern United States14. There is evidence that adaptive introgression occurred during their invasion21.
Methods
Sampling and sample information
We collected 12 diploid worker bees from the brood frames of each of 30 Africanized honeybee colonies from four apiaries all located at Iaras city, São Paulo, Brazil (Table 1), located approximately 200 km from Rio Claro. These colonies were obtained from natural swarms within the State of São Paulo where the Africanized honeybee invasion began. We performed a single DNA extraction for each colony by pooling ¼ of a thorax from each of the 12 workers22. We used the Mag-Bind Blood DNA kit (Omega Biotek Store) with the manufacturer's recommended protocol to extract an average of 4.71±1.42 μg of high-quality DNA from each of the colonies.
DNA samples (one pooled sample of 12 workers from each colony) were submitted to The Centre for Applied Genomics (Toronto, ON) for library preparation and high-throughput sequencing. In brief, DNA was quantified by Qubit HS assay and 200 ng of DNA was used as input material for the TruSeq Nano DNA Sample Preparation protocol (Illumina, Inc.) following Illumina's recommendation. DNA was sheared to 550-bp on average using a Covaris S2 system (Duty cycle: 10%; Intensity 2; Burst per second: 200; Treatment time: 44 s; Mode: Frequency sweeping). The sheared DNA was end-repaired and the 3' ends were adenylated prior to ligation of the TruSeq adapters. The library was enriched by PCR using different indexed adapters to allow for multiplex sequencing using the following conditions: 95 °C for 3 min followed by 8 cycles of 98 °C for 20 s, 60 °C for 15 s and 72 °C for 30 s, and finally an extension step at 72 °C for 5 min.
Final TruSeq Nano DNA genomic libraries were validated on a Bioanalyzer 2100 DNA High Sensitivity chip (Agilent Technologies) for size and by qPCR using the Kapa Library Quantification Illumina/ABI Prism Kit protocol (KAPA Biosystems) for quantities. Ten libraries were pooled in equimolar quantities and sequenced on a HiSeq 2500 platform on a high throughput flowcell with the Illumina TruSeq V4 sequencing chemistry following Illumina’s recommended protocol to generate paired-end reads of 150-bases in length.
Genome alignment and variant calling
Each colony’s sequenced reads (N=30 colonies) were trimmed of Illumina Adaptors using Trimmomatic v0.32 then aligned to the most recent version of the honeybee genome (Amel_4.5 (ref. 23)) using BWA aligner v0.7.5 (ref. 24). Paired alignments were then merged with SAMTOOLS v 0.1.19 (ref. 25) and re-aligned using STAMPY v1.0.21 (ref. 26) with divergence (--d) set at 0.02. We marked and removed duplicate reads with PICARD v 1.141 and re-aligned around indels using GATK IndelRealigner v 3.1 (ref. 27) (Fig. 1; Data Citation 1).

To identify all variants found within our samples, we used two independent variant callers (Fig. 1): VARSCAN28 v2.3.7 and GATK UnifiedGenotyper (Data Citations 2 and 3) We used GATK with --ploidy 2 to identify only the location of variants and were unconcerned with specific genotype calls. We called all variant sites in GATK and VARSCAN using default parameters. We removed indels and all SNPs within 10 bp of indels, removed all unmapped scaffolds (Scaffolds 17.XXX or GroupUn) and mitochondrial sequence (Scaffold 18.1), and removed SNPs of low quality (Q<25) or in areas of low genomic complexity, thus reducing the potential for calling erroneous SNPs due to paralogous sequence or misaligned reads3 (Fig. 1). We retained all SNPs that were identified using both variant callers and that passed our conservative filtering procedures, above. Because our data consist of pooled sequence for 30 colonies, we report the allele frequency at each site as called by VARSCAN in Variant Call Format (Data Citation 3).
SNP validation: Population differentiation and admixture
To quantify differentiation among contemporary Africanized populations in Brazil and ancestral honeybee populations, we used POPOOLATION2 v1.201 (ref. 29). We created a single input file containing our Africanized bee samples pooled into a single alignment as well as population-pooled alignments from ancestral honeybee populations from Africa (A lineage, N=11) and Europe (M lineage, N=9; C lineage N=9). The latter sequence data were obtained from a recent honeybee population genomics study3 and represents ancestral populations from which Africanized populations are derived. We generated a single MPILEUP file and extracted from it the 3,606,720 SNPs identified above in our Africanized honeybee samples. We estimated pairwise population differentiation on all sites with --min-count 6 --min-coverage 100 --max-coverage 800 --min-covered-fraction 0.8.
Code availability
We have not used any custom code and relied on previously available, validated, software packages; however, we have left our general pipeline available for re-use at the author’s GitHub (https://github.com/harpur/afz/blob/master/AHBPipeline.sh).
Data Records
We have curated a set of 3,606,720 SNPs identified in 360 Africanized honeybees across 30 colonies (Data Citations 2 and 3). The data consist of SNPs called across the most recent honeybee reference genome (Amel_4.5 (ref. 23)) in Variant Call File format on placed scaffolds. Because we utilized a pooled-sequencing method, all variant sites include the frequency of each alternate allele call for each colony. All sequence data are also available in BAM format (Data Citation 1; Table 1) allowing subsequent researchers to use updated SNP calling and genotype software when available.
Technical Validation
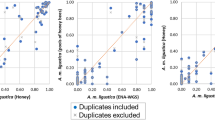
To validate that our samples are indeed Africanized and to confirm our SNP calls, we compared our current SNPs to those of a previous honeybee population genomics study that sequenced and analysed honeybee samples using similar methods as described herein3. Africanized bees are known to be derived from three of the major honeybee population groups: A, M, and C6,16. We found that 99.8% of the 3,606,720 SNPs found in AHBs, were also found within one or more of these ancestral populations. Africanized populations are expected to have higher A lineage ancestry relative to C and M lineage ancestry7,16. Using a regression model30, we demonstrated that allele frequencies within Africanized bees are more correlated with A lineage allele frequencies (GLM; r=0.529, P<2.2×10−16) relative to both M lineage allele frequency (r=0.102, P<2.2×10−16) and C lineage allele frequency (r=−0.08, P<2.2×10−16; Fig. 2), as we would expect from an Africanized population. As well, we find that AHB and A lineage are more similar genetically (Fst=0.02) than AHB versus M-lineage (0.04) and AHB versus C-lineage (0.05).

Red line shows results of linear model fit.
Additional Information
How to cite this article: Kadri, S. M. et al. A variant reference data set for the Africanized honeybee, Apis mellifera. Sci. Data 3:160097 doi: 10.1038/sdata.2016.97 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
References
Tarpy, D. R., Delaney, D. A. & Seeley, T. D. Mating frequencies of honey bee queens (Apis mellifera L.) in a population of feral colonies in the northeastern United States. PLos ONE 10, e0118734 (2015).
Wallberg, A. et al. A worldwide survey of genome sequence variation provides insight into the evolutionary history of the honeybee Apis mellifera. Nat Genet 46, 1081–1088 (2014).
Harpur, B. A. et al. Population genomics of the honey bee reveals strong signatures of positive selection on worker traits. P Natl Acad Sci USA 111, 2614–2619 (2014).
Garnery, L., Cornuet, J. M. & Solignac, M. Evolutionary history of the honey bee Apis mellifera inferred from mitochondrial DNA analysis. Mol Ecol 1, 145–154 (1992).
Arias, M. C. & Sheppard, W. S. Molecular phylogenetics of honey bee subspecies (Apis mellifera L.) inferred from mitochondrial DNA sequence. Molecular phylogenetics and evolution 5, 557–566 (1996).
Whitfield, C. W. et al. Thrice out of Africa: Ancient and recent expansions of the honey bee, Apis mellifera. Science 314, 642–645 (2006).
Ruttner, F . Biogeography and taxonomy of honeybees (Springer-Verlag, 1988).
Sheppard, W. S. A history of the introduction of honey bee races into the United States.2. Am Bee J 129, 664–667 (1989).
Sheppard, W. S. A history of the introduction of honey bee races into the United States.1. Am Bee J 129, 617–619 (1989).
Crane, E . The world history of beekeeping and honey hunting (Routledge, 1999).
Nogueira-Neto, P. The spread of a fierce African bee in Brazil. Bee World 45, 119–121 (1964).
Kerr, W. E. Introdução de abelhas africanas no Brasil. Brasil Apicola 3, 211–213 (1957).
Winston, M. L . The biology of the honey bee (Harvard University Press, 1987).
Winston, M. L . Killer bees: The Africanized honey bee in the Americas (Harvard University Press, 1992).
Weinstock, G. M. et al. Insights into social insects from the genome of the honeybee Apis mellifera. Nature 443, 931–949 (2006).
Harpur, B. A. et al. Assessing patterns of admixture and ancestry in canadian honey bees. Insect Soc 62, 479–489 (2015).
Harpur, B. A., Minaei, S., Kent, C. F. & Zayed, A. Management increases genetic diversity of honey bees via admixture. Mol Ecol 21, 4414–4421 (2012).
Munoz, I. et al. Reduced SNP panels for genetic identification and introgression analysis in the dark honey bee (Apis mellifera mellifera). PLos ONE 10, e0124365 (2015).
Chapman, N. C. et al. A SNP test to identify Africanized honeybees via proportion of ‘African’ ancestry. Mol Ecol Resour 15, 1346–1355 (2015).
Chandrasekaran, S. et al. Aggression is associated with aerobic glycolysis in the honey bee brain. Genes, brain, and behavior 14, 158–166 (2015).
Zayed, A. & Whitfield, C. W. A genome-wide signature of positive selection in ancient and recent invasive expansions of the honey bee Apis mellifera. P Natl Acad Sci USA 105, 3421–3426 (2008).
Ferretti, L., Ramos-Onsins, S. E. & Perez-Enciso, M. Population genomics from pool sequencing. Mol Ecol 22, 5561–5576 (2013).
Munoz-Torres, M. C. et al. Hymenoptera genome database: Integrated community resources for insect species of the order hymenoptera. Nucleic Acids Res 39, D658–D662 (2011).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595 (2010).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Lunter, G. & Goodson, M. Stampy: A statistical algorithm for sensitive and fast mapping of illumina sequence reads. Genome Res 21, 936–939 (2011).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43, 491–498 (2011).
Koboldt, D. C. et al. Varscan: Variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 25, 2283–2285 (2009).
Kofler, R., Pandey, R. V. & Schlotterer, C. Popoolation2: Identifying differentiation between populations using sequencing of pooled DNA samples (pool-seq). Bioinformatics 27, 3435–3436 (2011).
Chiang, C. W. K. et al. Rapid assessment of genetic ancestry in populations of unknown origin by genome-wide genotyping of pooled samples. PLoS genetics 6, e1000866 (2010).
Data Citations
NCBI SRA PRJNA324081 (2016)
NCBI dbSNP 1062539 (2016)
Harpur, B. A., Kadri, S.M, Orsi, R. O., & Zayed, A. Figshare https://doi.org/10.6084/m9.figshare.3501863 (2016)
Acknowledgements
The authors would like to thank Israel dos Santos for help in obtaining samples and for insightful discussions regarding Africanized bees in Brazil. This study was funded by a Discovery Grant from the Natural Sciences and Engineering Research Council of Canada (NSERC) and an Early Researcher Award from the Ontario Ministry of Research and Innovation (to A.Z.). B.A.H. was supported by an Elia Scholarship from York University and a NSERC Alexander Graham Bell CGS. S.M.K. was supported by a scholarship from Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP Process 2014/10150-2). We would also like to thank Juliana Sampson for the excellent support during the upload process to dbSNP.
Author information
Authors and Affiliations
Contributions
S.M.K. and B.A.H. contributed equally to this manuscript. B.A.H., S.M.K. and A.Z. designed the sampling scheme, S.M.K. collected samples, B.A.H. created the database, B.A.H., S.M.K., R.O.O. and A.Z. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
ISA-Tab metadata
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0 Metadata associated with this Data Descriptor is available at http://www.nature.com/sdata/ and is released under the CC0 waiver to maximize reuse.
About this article

Cite this article
Kadri, S., Harpur, B., Orsi, R. et al. A variant reference data set for the Africanized honeybee, Apis mellifera. Sci Data 3, 160097 (2016). https://doi.org/10.1038/sdata.2016.97
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/sdata.2016.97
This article is cited by
-
The honey bee genome-- what has it been good for?
Apidologie (2021)
