
Abstract
We present a structural data set of the 20 proteinogenic amino acids and their amino-methylated and acetylated (capped) dipeptides. Different protonation states of the backbone (uncharged and zwitterionic) were considered for the amino acids as well as varied side chain protonation states. Furthermore, we studied amino acids and dipeptides in complex with divalent cations (Ca2+, Ba2+, Sr2+, Cd2+, Pb2+, and Hg2+). The database covers the conformational hierarchies of 280 systems in a wide relative energy range of up to 4 eV (390 kJ/mol), summing up to a total of 45,892 stationary points on the respective potential-energy surfaces. All systems were calculated on equal first-principles footing, applying density-functional theory in the generalized gradient approximation corrected for long-range van der Waals interactions. We show good agreement to available experimental data for gas-phase ion affinities. Our curated data can be utilized, for example, for a wide comparison across chemical space of the building blocks of life, for the parametrization of protein force fields, and for the calculation of reference spectra for biophysical applications.
Design Type(s) | observation design • data integration objective |
Measurement Type(s) | protein conformation assessment |
Technology Type(s) | data representational model |
Factor Type(s) | amino acid • biochemical phenotype • peptidyl-amino acid modification |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others

Background & Summary
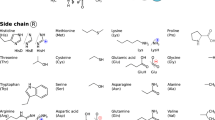
Proteins are the machinery of life. We here present a first-principles study of the conformational preferences of their basic building blocks—specifically, as summarized in Fig. 1: 20 proteinogenic amino acids and dipeptides, with different possible protonation states, and the conformational space changes resulting from attaching six divalent cations, i.e., Ca2+, Ba2+, Sr2+, Cd2+, Pb2+, and Hg2+. In past studies, a wide range of different approximate electronic structure methods has been applied to some of these proteinogenic amino acids—see, for example, references1–59. These studies have deepened our understanding of the conformational basics of individual building blocks, but a systematic comparison of properties of the different building blocks is complicated when relying on data from different sources. On the one hand this is due to the molecular models that may differ in protonation states and backbone capping. On the other, the simulations can differ in several ways:
-
Different sampling strategies or methods to generate conformers may have been used. Search-dependent settings, like energy cut-offs, can also have a significant impact on the results.
-
The levels of theory that have been applied range from semi-empirical to Hartree-Fock (HF) to density-functional theory (DFT) up to coupled-cluster calculations1–59.
-
Numerical settings, e.g., basis sets, can differ substantially and might lead to different results.

Top left and center: Schematic depiction of the backbone conformations of uncharged, zwitterionic, and dipeptide forms of the aminoacids considered in this work. Side chains are indicated by the letter R. Top right: Divalent ions considered for complexation with the 20 proteinogenic amino acids. Lower five rows: Side chains, including different protonation states where applicable, of the 20 proteinogenic amino acids considered in this work.
A further point that limits a quantitative comparison is the accessibility of the data from different studies. Energies, for example, often have to be extracted from table footnotes and/or the structural data is not always accessible in the Supplementary Information of the respective articles, sometimes even only accessible as figures in the manuscript. The data set presented here overcomes such limitations by covering a comprehensive segment of chemical space exhaustively, using a large scale computational effort. This study treats 20 proteinogenic amino acids, their dipeptides and their interactions with the divalent cations Ca2+, Ba2+, Sr2+, Cd2+, Pb2+, and Hg2+ (see Fig. 1 for an overview) on the same theoretical footing. The importance of peptide cation interactions may be highlighted by the fact that about 40% of all proteins bind cations60–62. Especially Ca2+ is important in a multitude of functions, ranging, for example, from blood clotting63 to cell signaling to bone growth64. Such calcium mediated functions can be disturbed by the presence of alternative divalent heavy metal cations like Pb2+, Cd2+, and Hg2+ (refs 62,65,66).
The conformations and total energies of each molecular system are calculated from first principles in the framework of density-functional theory (DFT)67,68 using the PBE generalized-gradient exchange-correlation functional69. Energies are corrected for van der Waals interactions using the Tkatchenko-Scheffler formalism70. In this formalism, pairwise C6[n]/r6 terms are computed and summed up for all pairs of atoms. r is the interatomic distance, a cut-off for short interatomic distances is applied, and C6[n] coefficients are obtained from the self-consistent electron density. The combined approach is referred to as ‘PBE+vdW’ throughout this work. This level of theory is robust for potential-energy surface (PES) sampling of peptide systems71–78. The curated data is provided as basis for comparative studies across chemical space to reveal conformational trends and energetic preferences. It can, for example, further be used for force-field development, theoretical studies at higher levels of theory, and as a starting point for theoretical calculations of spectra for biophysical applications.
Methods
Molecular models
This study covers a total of 280 molecular systems (summarized in Fig. 1). The number is the product of the following chemical degrees of freedom that were considered in our study:
20 proteinogenic amino acids. In case of (de)protonatable side chains, all protomers (different protonations states) were considered as well.
2 different backbone types, either free termini (considered in uncharged or zwitterionic form) or capped (N-terminally acetylated or C-terminally amino-methylated).
7 reflecting that the respective amino acid or dipeptide was considered either in isolation or with one of six different cation additions: Ca2+, Ba2+, Sr2+, Cd2+, Pb2+, or Hg2+.
Conformational search and energy functions
For the initial scan of the PES, the empirical force field OPLS-AA79 was employed, followed by DFT-PBE+vdW relaxations of the energy minima identified in the force field. The identified set of structures was then subjected to a further first-principles refinement step, ab initio replica-exchange molecular dynamics (REMD). An overview of the procedure is given in Fig. 2 and the steps are described in more detail below.

Schematic representation of the workflow employed to locate stationary points on the potential energy surfaces of the respective molecular systems.
Force-field based (OPLS-AA)79 global conformational searches (Step 1) were performed for all dipeptides and amino acids (i) without a coordinating cation and (ii) with Ca2+. These searches employed a basin hopping search strategy80,81 as implemented in the tool ‘scan’, distributed with the TINKER molecular simulation package82,83. We here use an in-house parallelized version of the TINKER scan utility that was first used in reference74. In this search strategy, input structures for relaxations are generated by projecting along normal modes starting from a local minimum. The number of search directions from a local minimum was set to 20. Conformers were accepted within a relative energy window of 100 kcal/mol and if they differ in energy from already found minima by at least 10−4 kcal/mol. The search terminates when the relaxations of input structures do not result in new minima.
After that, PBE+vdW relaxations (Step 2) were performed with the program FHI-aims84–86. FHI-aims employs numeric atom-centered orbital basis sets as described in reference 84 to discretize the Kohn-Sham orbitals. Different levels of computational defaults are available, distinguished by choice of the basis set, integration grids, and the order of the multipole expansion of the electrostatic (Hartree) potential of the electron density. For the chemical elements relevant to this work, ‘light’ settings include the so-called tier1 basis sets and were used for initial relaxations. ‘Tight’ settings include the larger tier2 basis sets and ensure converged conformational energy differences at a level of few meV (ref. 84). Unless noted otherwise, all energies discussed here are results of PBE+vdW calculations with a tier2 basis and ‘tight’ settings. Relativistic effects were taken into account by the so-called atomic zero-order regular approximation (atomic ZORA)87,88 as described in reference84. Previous comparisons to high-level quantum chemistry benchmark calculations at the coupled-cluster level, CCSD(T), demonstrated the reliability of this approach for polyalanine systems72,76, alanine, phenylalanine, and glycine containing tripeptides76, and alanine dipeptides with Li+ (ref. 73). Further benchmarks at the MP2 level of theory are reported below in the section Technical Validation.
The refinement (Step 3) by ab initio REMD89,90 is intended to alleviate the potential effects of conformational energy landscape differences between the force field and the DFT method. In REMD, multiple molecular dynamics trajectories of the same system are independently initialized and run in a range of different temperatures. Based on a Metropolis criterion, configurations are swapped between trajectories of neighboring temperatures. Thus, the simulations can overcome barriers and provide an enhanced conformational sampling in comparison to classical molecular dynamics (MD)90,91. The simulations were carried out employing a script-based REMD scheme that is provided with FHI-aims and that was first used in reference92. Computations were performed at the PBE+vdW level with ‘light’ computational settings. The run time for each REMD simulation was 20 ps with an integration time step of 1 fs. The frequent exchange attempts (every 0.04 or 0.1 ps) ensure efficient sampling of the potential-energy surface as shown by Sindhikara et al.93. The velocity-rescaling approach by Bussi et al.94 was used to sample the canonical distribution. Starting geometries for the replicas were taken from the lowest energy conformers resulting from the PBE+vdW relaxations in Step 2. REMD parameters for the individual systems, i.e. the number of replicas, acceptance rates for exchanges between replicas, the frequency for exchange attempts, and the temperature range, are summarized in Supplementary Table S1 of the Supplementary Information. Conformations were extracted from the REMD trajectories every 10th step, i.e. every 10 fs of simulation time. In order to generate a set of representative conformers, these structures were clustered using a k-means clustering algorithm95 with a cluster radius of 0.3 Å as provided by the MMSTB package96. The resulting arithmetic-mean structures from each cluster were then relaxed using PBE+vdW with ‘light’ computational settings. The obtained conformers were again clustered and cluster representatives were relaxed with PBE+vdW (‘tight’ computational settings) to obtain the final conformation hierarchies. The refinement step by REMD is essential, as shown in Fig. 3, which separately identifies the number of distinct conformers found in Step 2 and, subsequently, the number of additional conformers found in Step 3.

Blue segments of the bars and blue shaded numbers give the number of stationary points (‘conformers’) located in Step 2 of the search procedure detailed in Fig. 2. Red bar segments and red shading highlight the number of conformers that were additionally found during Step 3 of the search. The total number of conformers found for each system is the sum of the numbers found in steps two and steps three.
After step 2, a total of 17,381 stationary points was found for the amino acids and dipeptides in isolation and in complex with Ca2+. The refinement procedure in Step 3 increases this number to a total of 21,259 structures. Initial structures for the Ba2+, Cd2+, Hg2+, Pb2+ and Sr2+ binding amino acid and dipeptide systems were then obtained by replacing the Ca2+ cation in the amino acid and dipeptide structures binding a Ca2+ cation. These structures were subsequently relaxed with PBE+vdW employing ‘tight’ computational settings and a tier-2 basis set. This procedure results in 24,633 further conformers with bound cations. Altogether, we thus provide information on 45,892 stationary points of the PBE+vdW PES for all systems studied in this work.
The numbers of conformers identified in the searches are also given in Supplementary Table S2 of the Supplementary Information. Supplementary Tables S3 and S4 provide detailed accounts of how many structures were found for which amino acid/dipeptide in isolation or with attached cations.
Data Records
The curated data, consisting of the Cartesian coordinates of 45,892 stationary point geometries of the PBE+vdW PES (the main outcome of our work) and their potential energies computed at the ‘tight’/tier-2 level of accuracy in the FHI-aims code, is provided as plain text files sorted in directories (see Fig. 4). The PBE+vdW total energies are included since they are an integral part of the construction of our geometry data sets. Importantly, the stationary point geometries could be used as starting points to refine the total energy accuracy by higher-level methods, e.g., those discussed in ‘Technical Validation’ below. The folder structure is hierarchic and straightforward. The naming scheme is explained in the following:

Each folder, as exemplified for the Ca2+-coordinated cysteine dipeptide, contains coordinate files in two formats (standard XYZ and FHI-aims input), the computational settings file for FHI-aims (control.in), and the energy hierarchies (PBE+vdW, ‘tight’/tier-2 level) per system.
Description of the file types:
conformer.(...).xyz coordinates in standard xyz format in Å, readable by a wide range of molecule viewers, e.g. VMD97, Jmol (http://www.jmol.org/), etc.
conformer.(...).fhiaims coordinate file in FHI-aims geometry input format: for each atom of the particular system, the Cartesian coordinates are given in Å (atom [x] [y] [z] [element]). The electronic total energy (in eV) at the PBE+vdW level is given there as a comment.
control.in FHI-aims input file with technical parameters for the calculations. Please note that these files also include the exact specifications of the ‘tight’ numerical settings for all included elements.
hierarchy_PBE+vdW_tier-2.dat in each final subfolder, contains three columns: number of the conformer, total energy (in eV, PBE+vdW, tier-2 basis set, ‘tight’ numerical settings, computed with FHI-aims version 031011), and relative energy (in eV, relative to the respective global minimum).
The curated data is publicly available from two sources:
-
1
A website dedicated to this data set has been set up (http://aminoaciddb.rz-berlin.mpg.de) and allows users to browse and download the data and to visualize molecular structures online.
-
2
From the NOMAD repository (http://nomad-repository.eu) the data is available via the DOI 10.17172/NOMAD/20150526220502 [Data citation 1].
Technical Validation
The conformational coverage for the amino acid alanine is validated by comparing to a recent study by Maul et al.12. In that reference10, low energy conformers of alanine were reported, spanning an energy range of approximately 0.26 eV between the reported lowest and highest energy conformers. The level of theory used by Maul et al. was DFT in the generalized gradient approximation by means of the Perdew-Wang 1991 functional98. In our case, the force field based search step with subsequent PBE+vdW relaxations yields 5 conformers. The following ab initio REMD simulations increase the number of conformers to 15 within an energy range of 0.43 eV. The respective conformational energy hierarchies after global search and after REMD-refinement are shown in Fig. 5a. The results of our search (with the refinement step) are in good agreement with the data from reference12 that is also shown in Fig. 5a. Structures are shown in Fig. 5b. Nine of the ten conformers identified by Maul et al. can be confirmed. The single conformer that is missing (highlighted by an X in Fig. 5a) is not a stationary point of the PBE+vdW potential energy surface. Conformers 14 and 15 are classified as saddle points by analysis of the vibrational modes.

(a) The conformational energy hierarchies for alanine after the global search and the local refinement together with the reference hierarchy at the DFT-PW91 level that was published by Maul et al.12. Conformers indicated by black lines were found in the global search, the conformers in red were located only after the local refinement step. The blue line in the reference conformational hierarchy represents a minimum not found in our search and not present at the PBE+vdW level. (b) Conformations of the alanine molecule. Conformers marked with an asterisk (*) were found in the local refinement step of our search strategy. Atoms are color-coded as follows: Cyan (C), blue (N), red (O), white (H). The conformer labeled with X was found by Maul et al. in PW91 calculations12 but is unstable at the PBE+vdW level.
In order to further quantify the reliability of the DFT-PBE+vdW level of theory for peptides, beyond earlier benchmark work72,73,76 and especially with divalent cations, benchmark calculations were performed at the level of Møller-Plesset second-order perturbation theory (MP2)99,100 using the electronic structure program package ORCA101. Single-point energy calculations were performed for all fixed stationary-point DFT-PBE+vdW geometries in our data base for the amino acids alanine (Ala) and phenylalanine (Phe) with neutral N and C termini in isolation as well as in complex with a Ca2+ cation. Phe was selected to represent a ‘difficult’ example, i.e., the interaction of the cation with a larger aromatic side chain. The MP2 calculations did not include any frozen-core treatment (including semicore states is essential for Ca2+) and were performed using Dunning's correlation-consistent polarized core-valence basis sets (cc-pCVnZ), with n=T/Q/5 denoting the triple-zeta, quadruple-zeta, and quintuple-zeta basis sets respectively102. The calculated SCF (Hartree-Fock) and MP2 correlation energies were then individually extrapolated to the complete basis set (CBS) limit as follows: For SCF energies, we used the extrapolation strategy proposed by Karton and Martin103:
A, α, and the CBS-extrapolated energy are parameters determined from a least-squares fitting algorithm applied individually for each conformer. For the MP2 correlation energies, an extrapolation scheme proposed by Truhlar104 was applied:
Again, B, β, and the CBS-extrapolated energy are parameters determined from a least-squares fitting algorithm as before. A detailed account of all numbers is given in the Supplementary Information (Supplementary Table S5). Mean absolute errors between the density-functional approximation (DFA) relative energies and the basis-set extrapolated MP2 relative energies were calculated as follows:
where the index i runs over all N conformations of a given data set. ΔEi in principle denotes the energy difference between conformer i and the lowest-energy conformer of the set. The adjustable parameter c is used to shift the MP2 and DFA conformational hierarchies versus one another to obtain the lowest possible MAE, rendering the reported MAE value independent of the choice of any reference structure. Fig. 6a shows the corresponding obtained mean absolute errors (MAE) and maximal errors (maxi|ΔEiDFA− ΔEiMP2+c|) of different DFA calculations—performed with the FHI-aims code—with respect to benchmarks on the MP2 level obtained as described above. Within FHI-aims, the accuracy of integration grids and of the electrostatic potential was also verified by comparing ‘tight’ and ‘really_tight’ numerical settings, giving virtually identical results. The DFA level of theory of PBE+vdW shows a MAE well within chemical accuracy of for both structural sets of Ala and Phe; for Phe, the maximal error is . We next applied a different long-range dispersion treatment, a recent many-body dispersion model based on interacting quantum harmonic oscillators denoted as MBD*105, showing no significant improvement for the isolated amino acids. In line with ref. 76, applying the more expensive PBE0 (ref. 105) hybrid exchange correlation functional reduces the maximum deviation for Phe to ~57 meV, i.e., 1.3 kcal/mol. For Ala and Phe with neutral end caps in complex with a Ca2+ cation, Fig. 6b compares the same set of DFAs to MP2 benchmark energy hierarchies. However, obtaining basis-set converged total energies of the same accuracy as for the isolated peptides by straightforward CBS extrapolation proved remarkably more difficult when Ca2+ was involved. The reason is traced to the significant and slow-converging correlation contribution of the Ca2+ semicore electrons, which leads to large and conformation dependent basis set superposition errors (BSSE). This problem was verified for MP2 calculations in the FHI-aims and ORCA codes, with several different basis set prescriptions107, and for CCSD(T) calculations. Standard DFAs, if sufficiently accurate, have a significant advantage in this respect since they are not subject to comparable numerical convergence problems. To yet arrive at reliable CBS-extrapolated MP2 conformational energy differences, we thus subjected the SCF and correlation energies of each Ca2+ coordinated conformation to a counterpoise correction108,109 to minimize the effect of BSSE on the Ca2+ correlation energy contribution, prior to performing CBS extrapolation as described above. For the example of Ala+Ca2+ and assuming rigid conformers, the BSSE is estimated as:

Mean absolute error (MAE) and maximal error (in meV) between different relative energies at the DFA (PBE+vdW, PBE+MBD*, and PBE0+MBD*) and MP2 level of theory, using structures of obtained minima at the PBE+vdW level from the database for the systems of Ala and Phe with neutral end caps, both (a) in isolation and (b) in complex with a Ca2+ cation. Computational details are given in the text. Exact numbers are summarized in Table 1.
(Ala) represents the energy of Ala evaluated in the union of the basis sets on Ala and Ca2+, EAla(Ala) represents the energy of Ala evaluated in the basis set on Ala, etc. The individual BSSE errors are then subtracted from the SCF and correlation energy respectively. Phe+Ca2+ is treated equivalently. Complete numerical details are given in the Supplementary Information (Supplementary Table S6). Following this procedure, the MAE and maximal error values of various DFAs compared to MP2 are well within 1 kcal/mol for Ala+Ca2+. The PBE+vdW MAE for Phe+Ca2+ amounts to just above ~2 kcal/mol. The contributions from both the many-body dispersion and the hybrid PBE0 functional improve the MAE to just above 1 kcal/mol at to PBE0+MBD* level of theory. The maximum errors in the energy hierarchies between individual conformers are correspondingly larger. Overall, this assessment shows that our data base of conformer geometries constitutes, e.g., an excellent starting point for more exhaustive future benchmark work of new electronic structure methods for cation-peptide systems. For example, it would be very interesting to explore how F12 approaches, which address the correlation energy convergence problem explicitly, fare for a broad range of different Ca2+ containing conformations of our peptides.
As a final validation, we compare the correlation of calculated gas-phase amino acid-Ca2+ binding energies to the binding energy hierarchy found experimentally in a study by Ho et al.110. We calculate binding energy at the PES level as
Energies E denote the PBE+vdW Born-Oppenheimer potential energies, including Eamino acid of the lowest-energy conformers of the isolated amino acid and Ecomplex of the same amino acid in complex with a Ca2+ ion. Experimentally110, the gas-phase Ca2+ affinities of 18 proteinogenic amino acids were determined by fragmenting Ca2+ complexes with a combinatoric library of tripeptides at T≈330 K, recording the mass spectrometric peak intensities of different fragmentation products. Quantitative average relative binding energies of Ca2+ to different amino acids were thus inferred and can be compared to our findings, albeit with several important experiment-theory differences: (i) Entropy effects73,75,111 should affect the specific complexes probed experimentally but cannot be included into the calculated numbers in the exact same way, (ii) structural differences (e.g., protonation, dimerized amino acids) between the fragments recorded in experiment and the amino acids covered here, (iii) experimental Ca2+ affinities are not given for Asp and Glu because their gas-phase acidities, needed for data conversion, are not known. Fig. 7 compares the experimentally and theoretically inferred Ca2+ binding affinities qualitatively. The x-axis reflects the experimental binding affinity energy hierarchy, arranging amino acids from left to right in order of decreasing binding affinity. The y axis shows calculated binding energies according to equation (5). Perfect correlation of the experimental and calculated hierarchies would imply a strictly monotonic decrease of calculated Ebinding values from left to right. This monotonic trend is not obeyed exactly; however, in view of the significant differences (i) and (ii) above, the qualitative agreement is quite striking. Normalized correlation coefficients between the experimental (1) and calculated (2) binding affinity data were calculated following the formula:
with s12 being the covariance of data sets and si being the standard deviations of data sets i=1,2. The result, correlation coefficients of r12=0.979 or 0.909 for uncapped amino acids or dipeptides, respectively, also point to an overall remarkably good agreement. Finally, Fig. 7 also gives predicted Ebinding values for protonated (overall system charge +2) and deprotonated (overall system charge +1) Asp and Glu, reflecting the significant electrostatic attraction between cations and negatively charged (deprotonated) Asp and Glu side chains. The binding energy data sets are included as Supplementary Table S5.

The amino acids are ordered along the x axis from the highest to lowest experimental Ca2+ binding energy. Protonated and deprotonated Asp and Glu are not included among the experimental data and are here shown as predictions. Ebinding is high for deprotonated Asp and Glu since these forms of the amino acid would carry a negative charge.
Usage Notes
The present data contains stationary-point geometries (mainly minima, but also saddle points since no routine normal-mode analysis was performed) on the potential energy surface of the 20 proteinogenic amino acids and dipeptides, either isolated or in complex with a divalent cation (Ca2+, Ba2+, Sr2+, Cd2+, Pb2+, Hg2+). The users of this dataset may find openbabel112 (www.openbabel.org) to be a useful tool to convert FHI-aims and xyz files to other common file formats in chemistry.
Additional Information
How to cite this article: Ropo, M. et al. First-principles data set of 45,892 isolated and cation-coordinated conformers of 20 proteinogenic amino acids. Sci. Data 3:160009 doi: 10.1038/sdata.2016.9 (2016).
References
References
Yu, W. et al. Extensive conformational searches of 13 representative dipeptides and an efficient method for dipeptide structure determinations based on amino acid conformers. J. Comput. Chem. 30, 2105–2121 (2009).
Kishor, S., Dhayal, S., Mathur, M. & Ramaniah, L. M. Structural and energetic properties of α-amino acids: A first principles density functional study. Mol. Phys. 106, 2289–2300 (2008).
Császár, A. G. & Perczel, A. Ab initio characterization of building units in peptides and proteins. Prog. Biophys. Mol. Biol. 71, 243–309 (1999).
Bouchoux, G. Gas phase basicities of polyfunctional molecules. Part 3: Amino acids. Mass Spectrom. Rev. 31, 391–435 (2012).
Matta, C. F. & Bader, R. F. W. Atoms-in-molecules study of the genetically encoded amino acids. II. Computational study of molecular geometries. Proteins: Struct., Funct., Bioinf 48, 519–538 (2002).
Schlund, S., Muller, R., Grassmann, C. & Engels, B. Conformational analysis of arginine in gas phase-a strategy for scanning the potential energy surface effectively. J. Comput. Chem. 29, 407–415 (2008).
Császár, A. G. On the structures of free glycine and α-alanine. J. Mol. Struct. 346, 141–152 (1995).
Császár, A. G. Conformers of gaseous glycine. J. Am. Chem. Soc. 114, 9568–9575 (1992).
Riffet, V., Frison, G. & Bouchoux, G. Acid-base thermochemistry of gaseous oxygen and sulfur substituted amino acids (Ser, Thr, Cys, Met). Phys. Chem. Chem. Phys. 13, 18561–18580 (2011).
Kabelac, M., Hobza, P. & Spirko, V. The ab initio assigning of the vibrational probing modes of tryptophan: Linear shifting of approximate anharmonic frequencies vs. multiplicative scaling of harmonic frequencies. Phys. Chem. Chem. Phys. 11, 3921–3926 (2009).
Kaschner, R. & Hohl, D. Density functional theory and biomolecules: A study of glycine, alanine, and their oligopeptides. J. Phys. Chem. A 102, 5111–5116 (1998).
Maul, R., Ortmann, F., Preuss, M., Hannewald, K. & Bechstedt., F. DFT studies using supercells and projector-augmented waves for structure, energetics, and dynamics of glycine, alanine, and cysteine. J. Comput. Chem. 28, 1817–1833 (2007).
Selvarengan, P. & Kolandaivel, P. Potential energy surface study on glycine, alanine and their zwitterionic forms. J. Mol. Struct.: THEOCHEM 671, 77–86 (2004).
Cao, M., Newton, S. Q., Pranata, J. & Schafer, L. J. Mol. Struct. THEOCHEM 332, 251 (1995).
Jaeger, H. M., Schaefer, H. F., Demaison, J., Császár, A. G. & Allen, W. D. Lowest-lying conformers of alanine: Pushing theory to ascertain precise energetics and semiexperimental re structures. J. Chem. Theory Comput. 6, 3066–3078 (2010).
Beachy, M. D., Chasman, D., Murphy, R. B., Halgren, T. A. & Friesner, R. A. Accurate ab initio quantum chemical determination of the relative energetics of peptide conformations and assessment of empirical force fields. J. Am. Chem. Soc. 119, 5908–5920 (1997).
Baek, K. Y., Fujimura, Y., Hayashi, M., Lin, S. H. & Kim, S. K. Density functional theory study of conformation-dependent properties of neutral and radical cationic l-tyrosine and l-tryptophan. J. Phys. Chem. A 115, 9658–9668 (2011).
Chen, M. & Lin., Z. Ab initio studies of aspartic acid conformers in gas phase and in solution. J. Chem. Phys. 127, 154314 (2007).
Floris, F. M., Filippi, C. & Amovilli., C. A density functional and quantum monte carlo study of glutamic acid in vacuo and in a dielectric continuum medium. J. Chem. Phys. 137, 075102 (2012).
Heaton, A. L., Moision, R. M. & Armentrout, P. B. Experimental and theoretical studies of sodium cation interactions with the acidic amino acids and their amide derivatives. J. Phys. Chem. A 112, 3319–3327 (2008).
Armentrout, P.B., Gabriel, A. & Moision., R.M. An experimental and theoretical study of alkali metal cation/methionine interactions. Int. J. Mass Spectrom. 283, 56–68 (2009).
Nguyen, D. T. et al. A density functional study of the glycine molecule: Comparison with post-hartree-fock calculations and experiment. J. Comput. Chem. 18, 1609–1631 (1997).
Espinoza, C., Szczepanski, J., Vala, M. & Polfer, N. C. Glycine and its hydrated complexes: A matrix isolation infrared study. J. Phys. Chem. A 114, 5919–5927 (2010).
Boeckx, B., Nelissen, W. & Maes, G. Potential energy surface and matrix isolation ft-ir study of isoleucine. J. Phys. Chem. A 116, 3247–3258 (2012).
Close, D. M. Calculated vertical ionization energies of the common alpha-amino acids in the gas phase and in solution. J. Phys. Chem. A 115, 2900–2912 (2011).
Baek, K. Y., Hayashi, M., Fujimura, Y., Lin, S. H. & Kim, S. K. Investigation of conformation-dependent properties of l-phenylalanine in neutral and radical cations by using a density functional taking into account noncovalent interactions. J. Phys. Chem. A 114, 7583–7589 (2010).
Sahai, M. A. et al. First principle computational study on the full conformational space of l-proline diamides. J. Phys. Chem. A 109, 2660–2679 (2005).
Linder, R., Seefeld, K., Vavra, A. & Kleinermanns, K. Gas phase infrared spectra of nonaromatic amino acids. Chem. Phys. Lett. 453, 1–6 (2008).
Vyas, N. & Ojha, A. K. Investigation on transition states of [alanine+m2+] (m=ca, cu, and zn) complexes: A quantum chemical study. Int. J. Quant. Chem 112, 1526–1536 (2012).
Lavrich, R. J. et al. Experimental studies of peptide bonds: Identification of the c[sub 7][sup eq] conformation of the alanine dipeptide analog n-acetyl-alanine n[sup [prime]]-methylamide from torsion-rotation interactions. J. Chem. Phys. 118, 1253–1265 (2003).
Zhang, M., Huang, Z. & Lin, Z. Systematic ab initio studies of the conformers and conformational distribution of gas-phase tyrosine. J. Chem. Phys. 122, 134313 (2005).
Dokmaisrijan, S., Lee, V. S. & Nimmanpipug., P. The gas phase conformers and vibrational spectra of valine, leucine and isoleucine: An ab initio study. J. Mol. Struct.: THEOCHEM 953, 28–38 (2010).
Ceci, M. L. et al. Exploratory conformational analysis of n-acetyl-l-tryptophan-n-methylamide. an ab initio study. J. Mol. Struct.: THEOCHEM 631, 277–290 (2003).
Chen, M., Huang, Z. & Lin., Z. J. Mol. Struct. THEOCHEM 719, 153 (2005).
Rassolian, M., Chass, G. A., Setiadi, D. H. & Csizmadia., I. G. Asparagine-ab initio structural analyses. J. Mol. Struct.: {THEOCHEM} 666–667, 273–278 (2003).
Zamora, M. A. et al. An exploratory ab initio study of the full conformational space of n-acetyl-l-cysteine-n-methylamide. J. Mol. Struct.: {THEOCHEM} 540, 271–283 (2001).
Rai, A. K., Song, C. & Lin., Z. An exploration of conformational search of leucine molecule and their vibrational spectra in gas phase using ab initio methods. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 73, 865–870 (2009).
von Helden, G., Compagnon, I., Blom, M. N., Frankowski, M., Erlekam, U., Oomens, J., Brauer, B., Gerber, R. B. & Meijer, G. Mid-ir spectra of different conformers of phenylalanine in the gas phase. Phys. Chem. Chem. Phys. 10, 1248–1256 (2008).
Riffet, V. & Bouchoux, G. Gas-phase structures and thermochemistry of neutral histidine and its conjugated acid and base. Phys. Chem. Chem. Phys. 15, 6097–6106 (2013).
Shemesh, D., Sobolewski, A. L. & Domcke, W. Role of excited-state hydrogen detachment and hydrogen-transfer processes for the excited-state deactivation of an aromatic dipeptide: N-acetyl tryptophan methyl amide. Phys. Chem. Chem. Phys. 12, 4899–4905 (2010).
Gabor, P., Perczel, A., Vass, E., Magyarfalvi, G. & Tarczay., G. A matrix isolation study on ac-gly-nhme and ac-l-ala-nhme, the simplest chiral and achiral building blocks of peptides and proteins. Phys. Chem. Chem. Phys. 9, 4698–4708 (2007).
Bakker, J. M., Aleese, L. M., Meijer, G. & von Helden, G. Fingerprint ir spectroscopy to probe amino acid conformations in the gas phase. Phys. Rev. Lett. 91, 203003 (2003).
Blanco, S., Sanz, M. E., López, J. C. & Alonso, J. L. Revealing the multiple structures of serine. Proc. Natl. Acad. Sci. USA 104, 20183–20188 (2007).
Szidarovszky, T., Czakó, G. & Császár, A. Mol. Phys. 107, 761 (2009).
Boeckx, B. & Maes, G. Experimental and theoretical observation of different intramolecular h-bonds in lysine conformations. J. Phys. Chem. B 116, 12441–12449 (2012).
Meng, L. & Lin, Z. Comprehensive computational study of gas-phase conformations of neutral, protonated and deprotonated glutamic acids. Computational and Theoretical Chemistry 976, 42–50 (2011).
Shankar, R., Kolandaivel, P. & Senthilkumar, L. Interaction studies of cysteine with li+, na+, k+, be2+, mg2+, and ca2+ metal cation complexes. Journal of Physical Organic Chemistry 24, 553–567 (2011).
Fleming, G. J., McGill, P. R. & Idriss, H. Gas phase interaction of l-proline with be2+, mg2+ and ca2+ ions: a computational study. Journal of Physical Organic Chemistry 20, 1032–1042 (2007).
Hu, C.-H., Shen, M. & Schaefer., H. F. Glycine conformational analysis. J. Am. Chem. Soc. 115, 2923 (1993).
Barone, V., Biczysko, M., Bloino, J. & Puzzarini, C. Characterization of the elusive conformers of glycine from state-of-the-art structural, thermodynamic, and spectroscopic computations: Theory complements experiment. J. Chem. Theory Comput. 9, 1533–1547 (2013).
Ai, H. Q., Bu, Y. X., Li, P. & Zhang., C. The regulatory roles of metal ions (m+/2+=li+, na+, k+, be2+, mg2+, and ca2+) and water molecules in stabilizing the zwitterionic form of glycine derivatives. New J. Chem. 29, 1540–1548 (2005).
Baldauf, C. & Hofmann., H.-J. Ab initio mo theory-an important tool in foldamer research: Prediction of helices in oligomers of ω-amino acids. Helvetica Chimica Acta 95, 2348–2383 (2012).
Ramek, M., Kelterer, A.-M. & Nikolić, S. Ab initio and molecular mechanics conformational analysis of neutral l-proline. Int. J. Quant. Chem 65, 1033–1045 (1997).
Czinki, E. & Császár, A. G. Conformers of gaseous proline. Chem. Eur. J 9, 1008–1019 (2003).
Kang., Y. K. Ab initio molecular orbital calculations on low-energy conformers of N-Acetyl-N‘-methylprolineamide. J. Phys. Chem. 100, 11589 (1996).
Xu, S., Ke-Dong, W. & Peng-Fei, M. Conformation effects on the molecular orbitals of serine. Chinese Physics B 20, 33102 (2011).
Yuan, Y., Mills, M. J. L., Popelier, P. L. A. & Jensen, F. Comprehensive analysis of energy minima of the 20 natural amino acids. J. Phys. Chem. A 118, 7876–7891 (2014).
Karton, A., Yu, L.-J., Kesharwani, M. & Martin, J. M. L. Heats of formation of the amino acids re-examined by means of w1-f12 and w2-f12 theories. Theoretical Chemistry Accounts 133, 1483 (2014).
Kesharwani, M. K., Karton, A. & Martin, J. M. L. Benchmark ab initio conformational energies for the proteinogenic amino acids through explicitly correlated methods. assessment of density functional methods. J. Chem. Theory Comput. 12, 444–454 (2015).
Holm, R. H., Kennepohl, P. & Solomon, E. I. Structural and functional aspects of metal sites in biology. Chemical Reviews 96, 2239–2314 (1996).
Tainer, J. A., Roberts, V. A. & Getzoff, E. D. Protein metal-binding sites. Current Opinion in Biotechnology 3, 378–387 (1992).
Kirberger, M. & Yang, J. J. Structural differences between Pb2+- and Ca2+-binding sites in proteins: Implications with respect to toxicity. Journal of Inorganic Biochemistry 102, 1901–1909 (2008).
Zhou, M. et al. and Jianping Ding. A novel calcium-binding site of von Willebrand factor A2 domain regulates its cleavage by ADAMTS13. Blood 117, 4623–4631 (2011).
Cheng, R. & Zhorov, B. Docking of calcium ions in proteins with flexible side chains and deformable backbones. European Biophysics Journal 39, 825–838 (2010).
Sadiq, S., Ghazala, Z., Chowdhury, A. & Büsselberg, D. Metal toxicity at the synapse: Presynaptic, postsynaptic, and long-term effects. Journal of Toxicology 2012, 132671 (2012).
Sharma, S. K., Goloubinoff, P. & Christen, P. Heavy metal ions are potent inhibitors of protein folding. Biochemical and Biophysical Research Communications 372, 341–345 (2008).
Hohenberg, P. & Kohn, W. Inhomogeneous electron gas. Phys. Rev 136, B864–B871 (1964).
Kohn, W. & Sham, L. J. Self-consistent equations including exchange and correlation effects. Phys. Rev 140, A1133–A1138 (1965).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Tkatchenko, A. & Scheffler, M. Accurate molecular van der Waals interactions from ground-state electron density and free-atom reference data. Phys. Rev. Lett. 102, 073005 (2009).
Rossi, M. et al. Secondary structure of Ac-Alan-LysH+ polyalanine peptides (n=5,10,15) in vacuo: Helical or not? J. Phys. Chem. Lett. 1, 3465–3470 (2010).
Tkatchenko, A., Rossi, M., Blum, V., Ireta, J. & Scheffler, M. Unraveling the stability of polypeptide helices: Critical role of van der Waals interactions. Phys. Rev. Lett. 106, 118102 (2011).
Baldauf, C. et al. How cations change peptide structure. Chemistry-A European Journal 19, 11224–11234 (2013).
Chutia, S., Rossi, M. & Blum, V. Water adsorption at two unsolvated peptides with a protonated lysine residue: From self-solvation to solvation. J. Phys. Chem. B 116, 14788–14804 (2012).
Rossi, M., Scheffler, M. & Blum, V. Impact of vibrational entropy on the stability of unsolvated peptide helices with increasing length. J. Phys. Chem. B 117, 5574–5584 (2013).
Rossi, M., Chutia, S., Scheffler, M. & Blum, V. Validation challenge of density-functional theory for peptides-example of Ac-Phe-Ala5-LysH+. J. Phys. Chem. A 118, 7349–7359 (2014).
Schubert, F. et al. Native like helices in a specially designed [small beta] peptide in the gas phase. Phys. Chem. Chem. Phys. 17, 5376–5385 (2015).
Schubert, F. et al. Exploring the conformational preferences of 20-residue peptides in isolation: Ac-Ala19-Lys+H+ vs. Ac-Lys-Ala19+H+ and the current reach of DFT. Phys. Chem. Chem. Phys. 17, 7375–7385 (2015).
Jorgensen, W. L., Maxwell, D. S. & Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 118, 11225–11236 (1996).
Wales, D. J. & Doye, J. P. K. Global optimization by basin-hopping and the lowest energy structures of Lennard-Jones clusters containing up to 110 atoms. J. Phys. Chem. A 101, 5111–5116 (1997).
Wales, D. J. & Scheraga, H. A. Global optimization of clusters, crystals, and biomolecules. Science 285, 1368–1372 (1999).
Ponder, J. W. & Richards., F. M. An efficient Newton-like method for molecular mechanics energy minimization of large molecules. J. Comput. Chem. 8, 1016–1024 (1987).
Ren, P. & Ponder, J. W. Polarizable atomic multipole water model for molecular mechanics simulation. J. Phys. Chem. B 107, 5933–5947 (2003).
Blum, V. et al. Ab initio molecular simulations with numeric atom-centered orbitals. Computer Physics Communications 180, 2175–2196 (2009).
Havu, V., Blum, V., Havu, P. & Scheffler, M. Efficient integration for all-electron electronic structure calculation using numeric basis functions. Journal of Computational Physics 228, 8367–8379 (2009).
Ren, X. et al. Resolution-of-identity approach to Hartree-Fock, hybrid density functionals, RPA, MP2 and GW with numeric atom-centered orbital basis functions. New Journal of Physics 14, 053020 (2012).
van Lenthe, J. H., Faas, S. & Snijders, J. G. Gradients in the ab initio scalar zeroth-order regular approximation (ZORA) approach. Chem. Phys. Lett. 328, 107–112 (2000).
van Wullen, C. Molecular density functional calculations in the regular relativistic approximation: Method, application to coinage metal diatomics, hydrides, fluorides and chlorides, and comparison with first-order relativistic calculations. J. Chem. Phys. 109, 392–399 (1998).
Swendsen, R. H. & Wang, J.-S. Replica Monte Carlo simulation of spin-glasses. Phys. Rev. Lett. 57, 2607–2609 (1986).
Sugita, Y. & Okamoto, Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 314, 141–151 (1999).
Earl, D. J. & Deem, M. W. Parallel tempering: Theory, applications, and new perspectives. Phys. Chem. Chem. Phys. 7, 3910 (2005).
Beret, E. C., Ghiringhelli, L. M. & Scheffler, M. Free gold clusters: beyond the static, monostructure description. Faraday Discuss. 152, 153–167 (2011).
Sindhikara, D., Meng, Y. & Roitberg, A. E. Exchange frequency in replica exchange molecular dynamics. J. Chem. Phys. 128, 024103 (2008).
Bussi, G., Donadio, D. & Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 (2007).
Hartigan, J. A. & Wong, M. A. Algorithm as 136: A k-means clustering algorithm. Journal of the Royal Statistical Society. Series C (Applied Statistics) 28, 100–108 (1979).
Feig, M., Karanicolas, J. & Brooks III, C. L. MMTSB tool set: enhanced sampling and multiscale modeling methods for applications in structural biology. Journal of Molecular Graphics and Modelling 22, 377–395 (2004).
Humphrey, W., Dalke, A. & Schulten, K. VMD - Visual Molecular Dynamics. J. Molec. Graphics 14, 33–38 (1996).
Perdew., J. P. Unified Theory of Exchange and Correlation Beyond the Local Density Approximation. in Electronic Structure of Solids '91-Proceedings of the 75. WE-Heraeus-Seminar and 21st Annual International Symposium on Electronic Structure of Solids; Gaussig, Germany; 11–15 March 1991 (Akademie Verlag, Berlin, 1991).
Møller, C. & Plesset, M. S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev 46, 618–622 (1934).
Head-Gordon, M., Poplei, J. A. & Frisch., M. J. MP2 energy evaluation by direct methods. Chem. Phys. Lett. 153, 503–506 (1988).
Neese., F. The ORCA program system. WIREs Comput. Mol. Sci. 2, 73–78 (2012).
Woon, D. E. & Dunning, T. H. Gaussian basis sets for use in correlated molecular calculations. V. Corevalence basis sets for boron through neon. J. Chem. Phys. 103, 4572–4585 (1995).
Karton, A. & Martin, J. M. L. Comment on: "Estimating the Hartree-Fock limit from finite basis set calculations" [Jensen F (2005) Theor Chem Acc 113:267]. Theor. Chem. Acc. 115, 330–333 (2006).
Truhlar, D.G. Basis-set extrapolation. Chem. Phys. Lett. 294, 45–48 (1998).
Ambrosetti, A., Reilly, A. M., DiStasio, R. A. & Tkatchenko, A. Long-range correlation energy calculated from coupled atomic response functions. J. Chem. Phys. 140, 18A508 (2014).
Adamo, C. & Barone., V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 110, 6158–6170 (1999).
Zhang, I. Y., Ren, X., Rinke, P., Blum, V. & Scheffler, M. Numeric atom-centered-orbital basis sets with valence-correlation consistency from h to ar. New Journal of Physics 15, 123033 (2013).
Jansen, H. B. & Ros, P. Non-empirical molecular orbital calculations on the protonation of carbon monoxide. Chem. Phys. Lett. 3, 140–143 (1969).
Boys, S.F. & Bernardi., F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 19, 553–566 (1970).
Ho, Y.-P., Yang, M.-W., Chen, L.-T. & Yang, Y.-C. Relative calcium-binding strengths of amino acids determined using the kinetic method. Rapid Communications in Mass Spectrometry 21, 1083–1089 (2007).
Liwo, A., Khalili, M. & Scheraga., H. A. Ab initio simulations of protein-folding pathways by molecular dynamics with the united-residue model of polypeptide chains. Proc. Natl. Acad. Sci. USA 102, 2362–2367 (2005).
O'Boyle, N. et al. Open babel: An open chemical toolbox. Journal of Cheminformatics 3, 33 (2011).
Data Citations
Ropo, M., Baldauf, C., & Blum, V. NOMAD repository http://dx.doi.org/10.17172/NOMAD/20150526220502 (2015)
Acknowledgements
The authors are grateful to Matthias Scheffler (Fritz Haber Institute Berlin) for support of this work and stimulating discussions. Luca Ghiringhelli is gratefully acknowledged for his work on the script-based parallel-tempering scheme that is provided with FHI-aims and that was used in the present work. The authors thank Robert Maul and Karsten Hannewald for making available the original alanine geometries derived in their 2007 study for comparison with the present results. The authors further thank Mariana Rossi, Franziska Schubert, and Sucismita Chutia for sharing their extensive experience with all search methods employed in this work.
Author information
Authors and Affiliations
Contributions
M.R. performed the calculations to assemble all conformers. M.R. and C.B. curated the data. Validation calculations by DFAs and correlated methods other than PBE+vdW were carried out by M.S. M.R., C.B., V.B. designed the study and wrote the data descriptor.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
ISA-Tab metadata
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0 Metadata associated with this Data Descriptor is available at http://www.nature.com/sdata/ and is released under the CC0 waiver to maximize reuse.
About this article

Cite this article
Ropo, M., Schneider, M., Baldauf, C. et al. First-principles data set of 45,892 isolated and cation-coordinated conformers of 20 proteinogenic amino acids. Sci Data 3, 160009 (2016). https://doi.org/10.1038/sdata.2016.9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/sdata.2016.9
This article is cited by
-
Automatic characterization of drug/amino acid interactions by energy decomposition analysis
Theoretical Chemistry Accounts (2023)
-
Compressing local atomic neighbourhood descriptors
npj Computational Materials (2022)
-
Better force fields start with better data: A data set of cation dipeptide interactions
Scientific Data (2022)
-
TorsiFlex: an automatic generator of torsional conformers. Application to the twenty proteinogenic amino acids
Journal of Cheminformatics (2021)
-
Trapping lead in perovskite solar modules with abundant and low-cost cation-exchange resins
Nature Energy (2020)
