
Abstract
We report the first case of ocular myasthenia gravis (OMG) in a patient with complete tetraplegia, highlighting diagnostic and management challenges. Spinal multidisciplinary rural clinic and specialised inpatient Spinal Cord Injury Unit, NSW, Australia. A 61-year-old man with established C5 AIS A tetraplegia, presented with sudden onset of diplopia and bilateral ptosis, later diagnosed as OMG, in context of other complex co-morbidities, including a cervical cord syrinx, obstructive sleep apnoea and labile blood pressure. Clinical findings were consistent with fluctuating bilateral partial third and sixth nerve palsies. Acetylcholine receptor antibodies were negative, but electromyography demonstrated muscle fatigue. The ocular signs responded well to pyridostigmine. Medications taken before diagnosis, including solifenacin for neurogenic bladder overactivity, were ceased to avoid attenuating the anti-cholinesterase effect. However, the unopposed anti-cholinesterase activity led to frequent and painful abdominal spasms, associated with uncontrolled detrusor hyperreflexia and worsening autonomic dysreflexia (AD). A trans-vesical phenol block to treat this provided only short-lasting benefit. Pyridostigmine was ceased to avoid provoking his abdominal spasms and his regular medications were recommenced. It was decided that the most appropriate treatment for his distressing diplopia was an eye patch. After discharge home, he continued to experience problems with recurrent urinary tract infections, abdominal spasms, episodic postural hypotension and AD. After 5 months, the patient died from an acute myocardial infarction. This case report contributes new knowledge about the rare presentation of OMG in a person with chronic tetraplegia.
Similar content being viewed by others
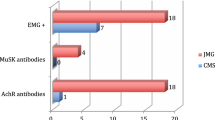

Myasthenia gravis (MG) is relatively rare, but the most common disorder of neuromuscular transmission, characterised by weakness and fatigability in skeletal muscles, including ocular, facial, bulbar, limb and respiratory muscle groups. Approximately 14% of all patients with MG present with isolated ocular symptoms as the only manifestation of their disease.1 However, ocular weakness may be present for more than 2 years before other muscle groups become involved. The clinical distinction between ocular MG (OMG) and an ocular presentation of MG therefore cannot always be made on clinical grounds alone. The circulating acetylcholine receptor antibody test also does not provide a specific distinction between the two groups, with acetylcholine receptor antibodies being detectable in approximately 50% of patients with OMG and 80–85% of patients with generalised MG.2 In any case, both groups whether sero-positive or sero-negative respond similarly well to immunosuppressant and anti-cholinesterase treatment.
In a person with established tetraplegia, pre-existing neurological impairment and other confounding spinal or age-related co-morbidities may mask typical clinical signs and symptoms of MG. Further, acetylcholinesterase inhibitors are first-line pharmacological therapy for MG, facilitating impulse transmission across the myoneural junction, that have opposing pharmacological actions to the anti-cholinergic medications commonly used to treat overactive neurogenic bladders in people with spinal cord injury (SCI).
The case reported below highlights potential challenges in diagnosing MG in a person with a high level SCI, as well as difficulties that may arise in managing common SCI-related impairments, such as bladder over activity and muscle spasticity because of conflicting medication effects.
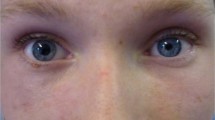
A 61-year-old man with C5 tetraplegia AIS A, as a result of falling from a bridge at the age of 22 years, presented to a multidisciplinary rural clinic conducted by the NSW Spinal Outreach Service for review. He reported sudden onset of right ptosis and diplopia for which his local medical officer had commenced antiplatelet therapy 4 weeks earlier on suspicion of brain stem vascular ischaemia. These symptoms developed on a background of unstable blood pressure (BP), with him experiencing symptomatic postural hypotension (with BP recorded as low as 60/40 mm Hg) as well as episodes of autonomic dysreflexia (with BP ranging up to 190/100 mm Hg). In addition, he had nocturnal hypertension associated with severe obstructive sleep apnoea, sub-optimally managed by non-invasive ventilation. Eight years prior, a ventriculo-peritoneal shunt had been inserted to treat a C3-7 cervical cord syrinx.
His regular medications include asprin 100 mg daily, clopidogrel 75 mg daily, baclofen 25 mg tds, solifenacin 5 mg nocte, diazepam 2.5 mg nocte, omeprazole 20 mg mane and bactrim DS daily (sulfamethoxazole 800 mg and trimethoprim 160 mg) for urinary prophylaxis and a bisacodyl enema.
At time of the clinic review, examination confirmed right-eye partial ptosis and weakness in abduction, with left-eye weakness in depression during adduction. There were no new neurological deficits in the cranial nerves, neck or upper limb muscles additional to his long standing tetraplegia. Given his past history of syringomyelia, contact was made with his treating neurosurgeon who thought his signs were consistent with internuclear ophthalmoplegia, warranting prompt transfer to Prince of Wales Hospital Spinal Unit to exclude shunt blockage with rostral extension of the cervical syrinx. Because of living in a rural region and the logistics of arranging an admission as well as difficulties with transport to Sydney, there was a delay in admission. Meanwhile, he developed a symptomatic urinary tract infection requiring admission to the local hospital, which ultimately enabled earlier inter-hospital transfer to Prince of Wales Hospital.
Examination on arrival at Prince of Wales Hospital showed sixth nerve lesions and a partial right third nerve lesion without evidence of pupillary abnormalities. Computerised tomography and magnetic resonance imaging scanning identified no change with regard to the size of the cervical syrinx and there were no other abnormalities involving the brain stem, brain or spinal cord. A neurological consultation was then sought. When assessed by the neurologist, the patient was found to have only right-sided ptosis and a complex pattern of impairment of ocular movement. Examination of the cranial nerves otherwise revealed no abnormalities. The pattern of impaired ocular movement on the right side suggested muscle rather than nerve involvement and a provisional diagnosis MG or OMG was made.
Serum acetylcholine receptor antibodies and muscle-specific kinase antibodies were negative. Electromyography demonstrated muscle fatigue on repeated stimulation and the patient responded well to a trial of pyridostigmine (30 mg), with complete resolution of the diplopia. Solifenacin, baclofen and diazepam were initially ceased because of potential anti-cholinergic and muscle relaxant properties, and regular pyridostigmine commenced.
Although the patient’s distressing diplopia was relieved, he unfortunately then experienced painful abdominal spasms associated with poorly controlled detrusor hyperreflexia, and worsening autonomic dysreflexia provoked by the pyridostigmine. Because MG being a relative contraindication for intra-vesical botulinum toxin, with the potential to precipitate a myasthenic crisis along with unpredictable therapeutic effects,3 a trans-vesical block using 6% aqueous phenol injections bilaterally into the region of the bladder neck was performed instead. This procedure provided some inhibition of bladder contractility with temporary relief of abdominal spasms and his symptoms of autonomic dysreflexia settled down. However, the response was not adequate, presumably because of an incomplete denervation of the bladder neck.
His abdominal pain was more distressing than the diplopia and it was difficult to achieve a satisfactory therapeutic balance and, therefore, it was decided that the most appropriate treatment was to manage the diplopia with eye patch and avoid provoking abdominal spasms.
The patient was recommenced on solifenacin, a selective anti-cholinergic medication, as well as diazepam prn to attempt to relieve detrusor overactivity and associated abdominal spasm pain. His obstructive sleep apnoea treatment was optimised. He was discharged home taking baclofen 25 mg tds for truncal spasticity, amlodipine 10 mg nocte for nocturnal hypertension, solifenacin 2.5 mg nocte and diazepam 2.5 mg as needed for abdominal spasms.
Post discharge, the patient continued to experience painful abdominal spasms, labile BP and recurrent urinary tract infections requiring multiple hospital admissions. Regular diazepam was commenced for refractory abdominal spasms. He died 5 months after diagnosis of MG from an unexpected acute myocardial infarction.
To our knowledge, this is the first report to describe a case of isolated OMG in a person with established complete tetraplegia. The authors did a literature search on PubMed, Medline and Google scholar. Other reported cases include a 54-year-old man with complete paraplegia, who was admitted for surgical repair of a stage-four sacral pressure ulcer and was noted to develop MG with symptoms of diplopia, fluctuating dysphagia and slurred speech.4 Two other reported cases involve a 92-year-old man with incomplete tetraplegia presenting with sudden dysphagia5 and a 45-year-old man with a 2-year history of stable chronic MG at the time of sustaining an acute traumatic injury with incomplete tetraplegia, in whom MG relapsed soon after discharge from inpatient rehabilitation secondary to sepsis.6
Diagnostic difficulties may arise where the pattern of weakness mimics impairment of either third, fourth or sixth cranial nerve function, unilaterally or bilaterally. Although the initial diagnosis was of a vascular event, it would be unusual for a brain stem ischaemic episode to result in the isolated symptom of diplopia. Similarly, the suggestion that acute onset of diplopia arose because of an upward extension of the cervical syrinx was also unlikely, in view of the absence of other brain stem symptoms and signs involving the cranial nerves and upper cervical segments. The variable pattern of ocular weakness also made this diagnosis very doubtful. In addition, there was also no indication of dissociated sensation which commonly coexists with an ascending syrinx.
The pattern of weakness which was noted at the time of the first neurological consultation pointed to specific muscle weakness rather than impairment of the ocular motor nerves. The diagnosis of myasthenic weakness was, therefore, probable on clinical grounds. Other possible diagnoses were the Lambert–Eaton syndrome, which is a paraneoplastic disorder and opthalmoplegia associated with hyperthyroidism. However, there were no clinical features to support either diagnosis.
The other striking feature of this case was the constant battle to balance symptomatic control of MG against competing pharmacological management of SCI-related impairments, such as detrusor over activity and truncal spasticity, with anti-cholinergic and anti-spasmodic medications.
Individuals with SCI experience higher mortality rates in comparison with an age-matched non-disabled population, particularly in those with tetraplegia and more complete lesions.7 Common causes of death in persons with SCI include cancer, ischaemic and non-ischaemic heart diseases and pneumonia. In our patient with a complete C5 tetraplegia almost 40 years post injury, acute myocardial infarction with cardiac arrest was the immediate cause of death, however, the underlying cause/s of death were likely to have been multifactorial. Both the increasing age at onset of MG and the high generalisation of OMG to MG, in the first 1–6 months after diagnosis,1 could have contributed to the demise of this patient. This may have been further complicated by his recurrent urinary tract infections, requiring hospital treatment with intravenous antibiotics, both of which can exacerbate MG. Ongoing lability in BP meant that his cardiovascular medications could not be optimised and this could have also contributed to worsening of his ischaemic heart disease. Finally, although his obstructive sleep apnoea was optimised before discharge, this may have worsened or compliance with therapy been compromised by his refractory abdominal spasms.
This case report highlights how spinal cord impairment with ageing and co-morbidities, such as syringomyelia, may delay diagnosis of MG as well as interact to confound treatment. In patients with established tetraplegia, the pre-existing neurological impairment may also mask typical patterns of skeletal muscle impairment in MG. The challenges experienced by the treating team in managing the opposing effects of anti-cholinesterase medication in patients with SCI requiring anti-cholinergic medication to treat symptoms associated with an overactive neurogenic bladder are clearly evident.
References
Grob D, Arsura EL, Brunner NG, Namba T . The course of myasthenia gravis and therapies affecting outcome. Ann N Y Acad Sci 1987; 505: 472–499.
Vincent A, Palace J, Hilton-Jones D . Myasthenia gravis. Lancet 2001; 357: 2122–2128.
Borodic G . Myasthenic crisis after botulinum toxin. Lancet 1998; 352: 1832.
Kolli S, Mathew KM, Thumbikat P, McClelland MR, Nair KP . Superimposed myasthenia gravis in chronic spinal cord injury: a case report. Spinal Cord 2011; 49: 1206–1207.
Kaux J-F, Ongena F, Wang F, Crielaard J-M, Foidart-Dessalle M . Sudden dysphagia in an elderly, quadriparetic patient. Ann Phys Rehabil Med 2009; 52: 59–65.
Lin C, Wang J, Wang Y, Pan S . Myasthenia gravis with superimposed spinal cord injury: a case report. J Rehabil Med 2008; 40: 684–686.
Middleton JW, Dayton A, Walsh J, Rutkowski SB, Leong G, Duong S . Life expectancy after spinal cord injury: a 50-year study. Spinal Cord 2012; 50: 803–811.
Acknowledgements
We thank Cathy Shorland BSW (Hons) for assisting with initial patient history.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Gounden, S., Lee, B., Mellick, R. et al. Ocular myasthenia gravis in a person with tetraplegia presenting challenges in diagnosis and management. Spinal Cord Ser Cases 2, 15037 (2016). https://doi.org/10.1038/scsandc.2015.37
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/scsandc.2015.37
