
Abstract
Study design:
Randomized, double-blind, placebo-controlled, two-period crossover.
Objectives:
To evaluate the efficacy and safety of arbaclofen placarbil (AP) in patients with spasticity secondary to spinal cord injury (SCI).
Setting:
United States and Canada.
Methods:
Patients received extended-release AP tablets 10, 20 or 30 mg every 12 h in one of two AP/placebo sequences, with 26 days of each treatment. The primary analysis compared Ashworth scale assessments of muscle tone between AP and placebo for the muscle group with maximum baseline Ashworth score. Secondary endpoints included a patient-rated Severity of Spasticity Scale.
Results:
In the primary analysis, AP significantly improved Ashworth scores compared with placebo over the dosing interval: least-squares mean reduction versus placebo was 0.60 for AP 20 mg (P=0.0059) and 0.88 for AP 30 mg (P=0.0007). The difference was significant for the pre-morning dose time point, 12 h after the prior evening dose, indicating that efficacy was maintained throughout the dosing interval. Treatment differences for AP 10 mg versus placebo were not significant. Severity of Spasticity ratings were significantly reduced for the combined 20/30-mg group versus placebo (P=0.018). No statistically significant differences between AP and placebo were observed for muscle strength. AP-related AEs were generally mild to moderate in intensity, and none led to early withdrawal or were serious.
Conclusion:
AP was well tolerated at all investigated dosages and, when administered at doses of 20 or 30 mg twice daily, was efficacious in reducing spasticity due to SCI.
Similar content being viewed by others

Introduction
Spasticity is a motor system disorder often associated with common neurologic disorders such as spinal cord injury (SCI), multiple sclerosis, stroke, cerebral palsy and traumatic brain injury.1 Up to 311 000 Americans are living with SCI,2 and 65–78% of patients with chronic SCI (persisting ⩾1 year after injury) have symptoms of spasticity.3 Further, more than 80% of people with multiple sclerosis have varying degrees of spasticity.4
Patients with spasticity experience a velocity-dependent increase in tonic stretch reflexes and an involuntary increase in muscle tone caused by structural lesions of the cerebrum, brainstem or spinal cord.5 Normal muscle-lengthening reactions occur in patients with early-stage spasticity. Over time, however, the amount of muscle stretch slowly decreases, resulting in the gradual development of musculoskeletal stiffness, atrophy, fibrosis and contracture. Spasticity can have a devastating effect on function, comfort and care delivery;3 therefore, it should be managed aggressively in its early stages to prevent permanent deformities and joint contracture.
Baclofen is a γ-aminobutyric-acid-B agonist6 that has been used in the United States for more than three decades7 to alleviate the signs and symptoms of spasticity resulting from SCI or multiple sclerosis.8 Oral racemic baclofen (R,S-baclofen) is approved for the treatment of spasticity;8 however, higher than approved doses are often used in clinical practice.9 Pharmacokinetic limitations of oral baclofen include a short duration of action, which necessitates t.i.d. or q.i.d. dosing, and night-time administration. Patients often forget mid-day doses of baclofen, indicating the potential value of an alternative extended-release medication. It has not been possible to develop an extended-release formulation of baclofen because its absorption is limited to the upper small intestine.7, 8 Further, oral baclofen is associated with adverse events (AEs) of the central nervous system (for example, somnolence, dizziness, weakness and confusion) and gastrointestinal system (for example, nausea).8 Rare but serious AEs include hallucinations, seizures and syncope.8 Intrathecal baclofen, which is used for the management of severe spasticity of a spinal cord or cortical origin,10, 11 can be associated with fewer systemic AEs than oral baclofen.3, 10
The antispasticity activity of baclofen resides in its R-isomer (R-baclofen).6 Arbaclofen placarbil (AP; XP19986; XenoPort, Santa Clara, CA, USA) is a novel, actively transported prodrug of R-baclofen that is administered in an extended-release oral formulation and is well absorbed throughout the gastrointestinal tract, including the colon.7 In preclinical studies, AP produced dose-proportional exposure to R-baclofen following oral dosing, with limited systemic exposure to intact prodrug.7 Compared with immediate-release baclofen, AP has a flatter pharmacokinetic profile and more sustained plasma levels allowing less frequent dosing and improved patient convenience. After single 20-mg oral doses of a prototype extended-release formulation of AP (SR1) in 10 healthy human volunteers, the time to maximal R-baclofen blood concentration (Tmax) was 5.05 h under fed conditions (unpublished data on file at XenoPort Inc.), which is longer than the comparable mean Tmax of 1.9 h after administration of racemic baclofen in a separate study.12 The associated reduction in fluctuations in blood levels may result in fewer AEs and enhanced efficacy when compared with immediate-release baclofen.
The objective of this study was to evaluate the efficacy and safety of a prototype extended-release formulation of AP (SR1) in the treatment of patients with spasticity secondary to SCI.
Materials and methods
Study design
This was a multiple-dose, randomized, double-blind, placebo-controlled, two-period crossover study (Figure 1a). Patients tapered off prohibited medications during a washout period and were required to be off antispasticity therapy for at least 24 h before the start of a 7-day, run-in baseline period, during which they received a placebo every 12 h (q12 h). The patients were then randomized to receive a prototype extended-release formulation of AP (SR1) 10, 20 or 30 mg q12 h (every morning and every evening) in one of two AP/placebo sequences. Patients were instructed to take all doses within 10 min after finishing a meal or snack. The patients received 26 days each of AP and placebo treatment. As described in Figure 1b, each treatment segment included 3–9 days of blinded titration. Higher target doses required correspondingly longer titration periods: 3 days for the target dose of 10 mg q12 h; 6 days for the target dose of 20 mg q12 h and 9 days for the target dose of 30 mg q12 h. The titration period was followed by ⩾1 week of treatment at the target dose, and then 3–9 days of blinded tapering from the target dose. Higher target doses required longer taper periods, corresponding to the length of the respective titration periods. The duration of treatment at the target dose was 7 days for the 30-mg q12 h dose. For lower doses, which had shorter titration and taper periods, time at target dosage was correspondingly longer. After tapering in the first treatment segment, a 3-day washout period occurred during which placebo was administered every 12 h prior to the titration period for the second treatment segment. The tapering period for the second treatment segment was followed by a 3-day placebo washout prior to a final assessment.

(a) Study design. (b) Placebo run-in, treatment and placebo washout schedule for patients randomized to receive AP 10, 20 or 30 mg. AP, arbaclofen placarbil.
Patients
Patients were eligible if they were diagnosed with spasticity secondary to SCI between C5 and T12, and were ⩾12 months post-injury with a stable neurologic deficit of traumatic or non-traumatic etiology, including resected, non-metastatic solid tumors intrinsic to or impinging upon the spinal cord, with no recurrence for ⩾5 years after resection. The eligible patients were 18–65 years old and had an Ashworth scale13 score of ⩾2 (0–4 scale) in at least one lower-limb muscle group at the baseline visit after washout of spasticity treatments. Although an active pain syndrome was allowed, chronic use of long-acting opioids at a dosage of >60 mg morphine equivalents per day for the treatment of pain was an exclusion criterion. Other key exclusion criteria were traumatic brain injury; cognitive deficit of any etiology; current anxiety disorder; depression or other psychiatric syndrome, active urinary tract infection or history of frequently recurrent urinary tract infections (>1 per month).
The study was conducted in accordance with good clinical practice guidelines, all applicable regulatory requirements and the guiding principles of the Declaration of Helsinki on biomedical research involving human subjects. Written informed consent was obtained from each subject before any study-related activities were performed. The protocol was approved by institutional review boards at the participating institutions, eight in the United States and two in Canada.
Screening assessments
Patients were assessed at the end of baseline using the American Spinal Cord Injury Association (ASIA) Impairment Scale,14, 15 the Functional Independence Measure (FIM)16 and the Patient Reported Impact of Spasticity Measure (PRISM)17 to characterize their disability. Scales were not used as treatment outcome measures because of treatment period brevity.
Outcome measures
The primary efficacy measure was the Ashworth scale assessment of muscle tone, which was obtained for six muscle groups on the most affected side of the body: hip abductors/adductors, knee flexors/extensors and ankle flexors/extensors. Five-point Ashworth scale scores ranged from 0 (no increase in tone) to 4 (affected part rigid in flexion or extension).13 Scores were assessed at baseline and four times (before and 2, 4 and 6 h after the morning dose) on day 17 of dosing in each treatment period, when patients had been at their target dose for at least a week. Dosing and assessment times were consistent throughout the study. Ashworth assessors were trained to a standardized method, using a videotape prepared by one of the authors (PN). The primary endpoint was the difference in Ashworth scores between AP and placebo on day 17 of dosing for the muscle group(s) with the maximum baseline Ashworth score.
A logpad/electronic diary was used twice daily between study visits, through the final visit, to record the severity of spasticity, pain owing to spasticity and sleep and dosing information. Weekly average score on the patient-rated Severity of Spasticity Scale18 and weekly average severity of pain due to muscle spasms were secondary efficacy measures. Severity of spasticity was rated by patients twice daily, after awakening each morning (regarding the previous night) and before retiring each evening (regarding the current day), using a four-point scale (0=none, 1=mild, 2=moderate and 3=severe) in response to the question ‘How would you rate the severity of your spasticity in general?’ Patients were asked to rate ‘severity of pain due to your muscle spasms’ twice daily on a numerical scale ranging from 0 (no pain due to muscle spasms) to 10 (the most severe pain imaginable due to muscle spasms). Other secondary efficacy measures were average Ashworth score (for all muscle groups), patellar and Achilles deep tendon reflex scores, muscle strength score, Epworth Sleepiness Scale19 score, night-time awakenings caused by muscle spasms, visual analogue scale (VAS) scores for sleep quality, mood on final awakening and alertness on final awakening. AEs were recorded at each study visit, but there was no query for specific AEs. Urinalysis and blood samples for hematology and chemistry were collected at each study visit.
Statistical analyses
The intent-to-treat (ITT) population included all patients randomized during double-blind treatment. The full analysis population, which was used for efficacy analyses, was defined as the subset of the ITT population who had no major entry criteria violations, took ⩾1 dose of scheduled study drug and had ⩾1 primary efficacy endpoint measurement (using the Ashworth scale) after randomization. The safety population included all patients who received ⩾1 dose of study drug.
Primary analysis of the difference in Ashworth scores between AP and placebo used a repeated-measures analysis of variance model with fixed effects for cohort, treatment sequence, time, treatment sequence-by-time and cohort-by-time interaction. Supportive analyses for the primary efficacy endpoint comprised Wilcoxon signed rank test for comparison of AP doses with placebo and an ANCOVA model for the maximum Ashworth score.
Comparisons of the combined 20/30-mg AP and placebo groups and of the 30-mg AP and placebo groups were performed simultaneously using the Hochberg procedure. Statistical significance was shown if P-values from both tests were <0.05 or the smallest P-value was <0.025 if the larger P-value was >0.05. The 20-mg AP and placebo groups were compared when comparison of the combined 20/30-mg and placebo groups was statistically significant. The 10-mg AP and placebo groups were to be compared if comparison of the 20-mg AP and placebo groups was statistically significant. Analysis of secondary efficacy endpoints used the same model used for primary analysis of the primary endpoint.
Safety evaluation was based on the proportion of patients with AEs, serious AEs, treatment-related AEs, AEs of various levels of intensity and AEs leading to withdrawal. Changes from baseline were assessed for other safety assessments.
Results
Patient disposition
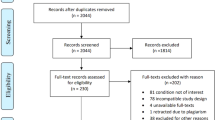
The ITT and safety populations included 37 patients: 10 randomized to receive AP 10 mg b.i.d., 13 to receive AP 20 mg b.i.d. and 14 to receive AP 30 mg b.i.d. (Figure 2). The full analysis population comprised 35 patients who completed both treatment sequences and completed the study. Two patients discontinued prematurely because of protocol deviations.

Patient disposition. AP, arbaclofen placarbil; ITT, intent to treat.
Demographic and baseline characteristics
Patient demographic and baseline characteristics were generally comparable among treatment groups (Table 1). Most patients were male (70.3%) and White (67.6%); median age ranged from 35 to 43 years, and the majority of patients (91.9%) had a traumatic SCI etiology. The mean maximum Ashworth score (for the muscle group with the highest baseline score) was 3.1. The average Ashworth score for the six muscle groups on the most affected side of the body had a mean of 1.9. Ashworth scores were comparable across dosage groups. The mean muscle strength scores ranged from 1.3 to 1.9 across the six muscle groups. The mean patellar reflex and Achilles reflex scores were both 2.9. The mean weekly average Severity of Spasticity score was 1.6. Overall, 40.5% of the patients had an ASIA-A classification. Overall, 51.4% of the patients previously received baclofen or tizanidine, which was washed out prior to baseline.
Primary efficacy endpoint
Treatment with AP 20 and 30 mg significantly improved spasticity compared with placebo over the pre-dose and 2-, 4- and 6-h post-dose time points as measured by the difference in the maximum Ashworth score on day 17 in the full analysis population (Figure 3). The least-squares mean reduction versus placebo was 0.60 for AP 20 mg (P=0.0059) and 0.88 for AP 30 mg (P=0.0007). By contrast, treatment response to AP 10 mg did not differ significantly from placebo.

Reduction in maximum Ashworth scores with AP treatment. For the respective dose groups, each bar represents the mean difference in maximum Ashworth score between AP and placebo treatments at the corresponding time points in relation to the morning dose at the end of the target dosing periods shown in Figure 1. Larger negative differences correspond to greater reductions in Ashworth score on AP compared with placebo. *P<0.05; †P<0.01. AP, arbaclofen placarbil.
Results for the pre-morning dose time point, representing the 12 h after the prior evening's dose, indicated that efficacy was maintained throughout the dosing interval and was dose-dependent. Compared with placebo, significant reductions in maximum Ashworth score were noted for AP 20 mg (P=0.0097) and 30 mg (P=0.0044) (Figure 3). Supportive analyses showed similar results.
Secondary efficacy endpoints
The score for patient-rated Severity of Spasticity was significantly reduced for the combined AP 20/30-mg group compared with placebo (least-squares mean, −0.24; P=0.018) (Figure 4). Compared with placebo, the average Ashworth score (average Ashworth score obtained on day 17 of dosing across the six muscle groups) was significantly reduced in the 20- (P=0.0015) and 30-mg (P=0.0043) AP groups. The mean Achilles reflex scores were significantly lower in the 30-mg AP group compared with placebo before (−0.7; P=0.0313) and 4 h after (−0.8; P=0.0156) the morning dose. No statistically significant differences between AP and placebo were observed for patellar reflex score or muscle strength scale scores; weekly frequency of night-time awakenings caused by muscle spasms; severity of pain due to spasms; Epworth Sleepiness Scale score or VAS scores for sleep quality, mood on final awakening or alertness on final awakening.

Decrease in severity of spasticity after AP treatment. For the respective dose groups, each bar represents the mean difference in Severity of Spasticity score between AP and placebo treatments during the target dosing periods shown in Figure 1. Larger negative differences correspond to greater reductions in Severity of Spasticity score on AP compared with placebo. *P=0.018, combined 20-mg b.i.d. and 30-mg b.i.d. groups versus placebo. AP, arbaclofen placarbil.
Safety
Overall, AP was well tolerated at all doses. No deaths, treatment-related serious AEs or discontinuations because of an AE were reported. In this crossover study, 75.7% of the patients reported at least one treatment-emergent AE when receiving AP and 48.6% of the patients reported at least one treatment-emergent AE when receiving placebo. The most frequently reported treatment-emergent AEs in the combined AP group were urinary tract infection (10.8%), pain in extremity (8.1%), insomnia (8.1%) and nasopharyngitis (8.1%) (Table 2). AEs typically associated with baclofen treatment were infrequent: dizziness (5.4%), somnolence (2.7%), weakness (2.7%) and confusion (0%). Treatment-emergent AEs were generally of mild-to-moderate intensity. Only one event of pain in extremity (2.7%) was considered by the investigator to be related to study drug. No clinically significant abnormalities in vital signs or laboratory or ECG results were observed during treatment with AP.
Discussion
In this study of patients with spasticity due to SCI, treatment with AP 20 and 30 mg b.i.d. doses resulted in statistically significant reductions in maximum Ashworth scale score compared with placebo at all time points in relation to dosing. The significant effect at the pre-morning dose time point, which was 12 h after the prior evening dose, indicates that efficacy was maintained throughout the dosing interval. The efficacy of AP was also shown by reductions compared with placebo in patient-rated, weekly average Severity of Spasticity scores in the combined 20- and 30-mg b.i.d. dose groups.
AP was well tolerated at all evaluated doses. No patient withdrew because of an AE, and AEs were generally mild to moderate in intensity. Importantly, AP was associated with a low incidence of AEs typically associated with oral baclofen treatment, including dizziness (5.4%), somnolence (2.7%) and weakness (2.7%), and no instances of confusion were reported.
These results are compelling in view of the challenges associated with demonstrating efficacy in this patient population.15 The level of improvement associated with AP 20 and 30 mg b.i.d. in this study is comparable to that reported in other studies with oral baclofen20 and tizanidine.21 In comparison with the published data regarding those treatments, AP in the present study showed a generally favorable tolerability profile. Although a lower incidence of systemic AEs has been reported with intrathecal than with oral baclofen, this advantage is counterbalanced by increased costs and safety issues associated with the use of an implantable pump.10 The recent approval of onabotulimtoxin-A (Botox) for the treatment of upper-limb spasticity in adults has provided an effective new approach to the management of spasticity for patients with focal spasticity.22 However, onabotulimtoxin-A may cause marked weakness of the injected muscles.1, 22 Less frequently, its effects may spread from the area of injection, resulting in generalized muscle weakness and other potentially serious AEs.
This study has some limitations. The population was small (n=37) and the duration of treatment (including initial titration and end-of-treatment taper) was 26 days. No patient global assessment or quality-of-life (QOL) measure was included as an outcome measure because it was expected that changes on such measures may not occur over the short treatment duration of this study. A global or QOL outcome would be particularly important for ASIA-C and D patients who may rely on spasticity for more activities of daily living. Although treatment sequence effect or period effect are often the problems in crossover studies, treatment sequence effect was not detected through the primary analysis model, and period effect was not detected through the supportive analysis model.
In conclusion, this randomized, double-blind, placebo-controlled, two-period crossover study showed that AP, administered at doses of 20 or 30 mg twice daily, is efficacious in alleviating spasticity due to SCI. AP had a durable treatment effect over the 12-h dosing interval, as shown by a significant reduction in maximum Ashworth Scale score pre-dose compared with placebo. AP significantly reduced the subject-rated severity of spasticity for the combined AP 20/30-mg dose groups compared with placebo. Furthermore, AP was well tolerated at all dosages investigated in the study.
References
Elbasiouny SM, Moroz D, Bakr MM, Mushahwar VK . Management of spasticity after spinal cord injury: current techniques and future directions. Neurorehabil Neural Repair 2010; 24: 23–33.
National SCI Statistical Center. National Spinal Cord Injury Statistical Center. Spinal cord injury facts and figures at a glance. The National SCI Statistical Center Web Site. Available at https://www.nscisc.uab.edu/public_content/pdf/Facts%20and%20Figures%20at%20a%20Glance%202010.pdf. Accessed November 15, 2010.
Adams MM, Hicks AL . Spasticity after spinal cord injury. Spinal Cord 2005; 43: 577–586.
Rizzo MA, Hadjimichael OC, Preiningerova J, Vollmer TL . Prevalence and treatment of spasticity reported by multiple sclerosis patients. Mult Scler 2004; 10: 589–595.
Young RR . Spasticity: a review. Neurology 1994; 44: S12–S20.
Misgeld U, Bijak M, Jarolimek W . A physiological role for GABAB receptors and the effects of baclofen in the mammalian central nervous system. Prog Neurobiol 1995; 46: 423–462.
Lal R, Sukbuntherng J, Tai EH, Upadhyay S, Yao F, Warren MS et al. Arbaclofen placarbil, a novel R-baclofen prodrug: improved absorption, distribution, metabolism, and elimination properties compared with R-baclofen. J Pharmacol Exp Ther 2009; 330: 911–921.
Schwarz Pharma. Kemstro™ (Baclofen Orally Disintegrating Tablets) (Prescribing Information). Schwarz Pharma: Milwaukee, WI, 2004.
Smith CR, LaRocca NG, Giesser BS, Scheinberg LC . High-dose oral baclofen: experience in patients with multiple sclerosis. Neurology 1991; 41: 1829–1831.
Medtronic Inc. Lioresal® Intrathecal (Baclofen Injection) (Prescribing Information). Medtronic Inc: Minneapolis, MI, 2010.
Albright AL, Barry MJ, Fasick MP, Janosky J . Effects of continuous intrathecal baclofen infusion and selective posterior rhizotomy on upper extremity spasticity. Pediatr Neurosurg 1995; 23: 82–85.
Wuis EW, Dirks MJ, Termond EF, Vree TB, Van der Kleijn E . Plasma and urinary excretion kinetics of oral baclofen in healthy subjects. Eur J Clin Pharmacol 1989; 37: 181–184.
Ashworth B . Preliminary trial of carisoprodol in multiple sclerosis. Practitioner 1964; 192: 540–542.
Ditunno Jr JF, Young W, Donovan WH, Creasey G . The international standards booklet for neurological and functional classification of spinal cord injury. American Spinal Injury Association. Paraplegia 1994; 32: 70–80.
Fawcett JW, Curt A, Steeves JD, Coleman WP, Tuszynski MH, Lammertse D et al. Guidelines for the conduct of clinical trials for spinal cord injury as developed by the ICCP panel: spontaneous recovery after spinal cord injury and statistical power needed for therapeutic clinical trials. Spinal Cord 2007; 45: 190–205.
Linacre JM, Heinemann AW, Wright BD, Granger CV, Hamilton BB . The structure and stability of the Functional Independence Measure. Arch Phys Med Rehabil 1994; 75: 127–132.
Cook KF, Teal CR, Engebretson JC, Hart KA, Mahoney JS, Robinson-Whelen S et al. Development and validation of Patient Reported Impact of Spasticity Measure (PRISM). J Rehabil Res Dev 2007; 44: 363–371.
Lechner HE, Frotzler A, Eser P . Relationship between self- and clinically rated spasticity in spinal cord injury. Arch Phys Med Rehabil 2006; 87: 15–19.
Johns MW . A new method for measuring daytime sleepiness: the Epworth sleepiness scale. Sleep 1991; 14: 540–545.
Hudgson P, Weightman D . Baclofen in the treatment of spasticity. BMJ 1971; 4: 15–17.
Nance PW, Bugaresti J, Shellenberger K, Sheremata W, Martinez-Arizala A . Efficacy and safety of tizanidine in the treatment of spasticity in patients with spinal cord injury. North American Tizanidine Study Group. Neurology 1994; 44: S44–S51.
Allergan Inc. Botox (OnabotulinumtoxinA) (Prescribing Information). Allergan Inc.: Irvine, CA, 2010.
Acknowledgements
We acknowledge the contributions of the following investigators, in addition to Dr Ayyoub, who enrolled the study patients: David F Apple, MD, Karen Ethans, MD, Sunil Hegde, MD, Amit Jha, MD, Ki H (Alex) Kim, MD, Maureen D Miner, MD, Edward C Nieshoff, MD, Colleen O’Connell, MD, and Atul Patel, MD. This study was funded by XenoPort Inc. Writing support was provided by XenoPort Inc.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
F Jacob Huff, Dan Chen, Amy Bian and David Stamler are employees of XenoPort Inc. Patricia W Nance and Alberto Martinez-Arizala have received compensation for consulting for XenoPort Inc. Ziyad Ayyoub received research support from XenoPort in relation to this study.
Additional information
Presented at the American Academy of Neurology 2010 Annual Meeting, April 10–17, 2010, Toronto, Ontario, Canada.
Rights and permissions
About this article
Cite this article
Nance, P., Huff, F., Martinez-Arizala, A. et al. Efficacy and safety study of arbaclofen placarbil in patients with spasticity due to spinal cord injury. Spinal Cord 49, 974–980 (2011). https://doi.org/10.1038/sc.2011.43
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sc.2011.43
Keywords
This article is cited by
-
A perspective on molecular signalling dysfunction, its clinical relevance and therapeutics in autism spectrum disorder
Experimental Brain Research (2022)
