
Abstract
Technologies for the creation of topological carbon nanostructures have greatly advanced synthetic organic chemistry and materials science. Although simple molecular nanocarbons with a belt topology have been constructed, analogous carbon nanobelts with a twist—more specifically, Möbius carbon nanobelts (MCNBs)—have not yet been synthesized owing to their high intrinsic strain. Here we report the synthesis, isolation and characterization of a MCNB. Calculations of strain energies suggest that large MCNBs are synthetically accessible. Designing a macrocyclic precursor with an odd number of repeat units led to a successful synthetic route via Z-selective Wittig reactions and nickel-mediated intramolecular homocoupling reactions, which yielded (25,25)MCNB over 14 steps. NMR spectroscopy and theoretical calculations reveal that the twist moiety of the Möbius band moves quickly around the MCNB molecule in solution. The topological chirality that originates from the Möbius structure was confirmed experimentally using chiral HPLC separation and circular dichroism spectroscopy.

Similar content being viewed by others

Main
Organic chemists have striven to realize a wide variety of structural features on the molecular scale in nanocarbons. For example, nanocarbons with spherical, sheet-like, cylindrical or other exotic structures are usually obtained as mixtures on applying a high energy to the appropriate carbon sources1,2,3,4. In this context, molecular nanocarbon science, with which such substructures are created in a precisely controlled fashion, has attracted substantial attention, given that this approach is fundamentally able to circumvent the problem of the formation of such nanocarbon mixtures5,6.
The history of the development of molecular nanocarbon science can be classified in terms of topology. Cycloparaphenylene, which was first proposed in the 1930s, is a ring-shaped molecular nanocarbon that represents a partial structure of carbon nanotubes7,8,9,10. Although cycloparaphenylenes were initially difficult to synthesize due to their high intrinsic strain energies, several synthetic methods, such as those reported since 2008 by Jasti, Itami and Yamago and their co-workers11,12,13, have enabled the creation of molecular nanocarbons that exhibit non-trivial topologies, such as cages14,15,16,17, catenanes and knots18,19. The next breakthrough in this research field was the synthesis of a carbon nanobelt (CNB) achieved by our group in 201720. The fully fused, belt-shaped topology of CNBs creates two non-convertible faces, that is, the inner and outer faces. Since then, the synthetic chemistry of CNBs and related belt-shaped arenes has been intensively investigated worldwide in the context of the bottom-up construction of carbon nanotube substructures21,22 as well as in the creation of new functional molecular nanocarbon materials.
The CNB structural feature of irreducible inner and outer faces can be extended to aromatic molecules with the topology of a Möbius strip, which is the simplest example of a non-orientable surface (Fig. 1a). Möbius-type molecules are found in nature23 and have been of interest in synthetic organic chemistry, as demonstrated by the successful preparation of a non-conjugated Möbius-type double-stranded molecule by Walba et al. in 1982 (Fig. 1b, left)24. Aromatic single-stranded molecules with Möbius aromaticity were realized by Herges and co-workers in 200325, and Möbius aromaticity was further investigated by Grażyński and co-workers26 and Osuka and co-workers27,28. Despite several examples of single-stranded Möbius molecules29,30,31,32,33, double-stranded aromatic molecules with a Möbius topology still remain limited due to the difficulties associated with their synthesis. As shown in Fig. 1b, saturated linkers (–CH2O–) or chalcogen atom linkers (–S–) are necessary to reduce the strain caused by the Möbius topology (Fig. 1b, centre and right)34,35. Even though Möbius-type CNBs have been theoretically proposed since the 1990s36,37,38, a synthetic methodology to introduce Möbius topology to fully fused and fully conjugated all-sp2 carbon structures still remains to be developed in molecular nanocarbon science.

a, The models of belt- and Möbius-type strips. b, Previously reported belt-shaped double-stranded molecules with Möbius topology: a non-conjugated Möbius-type double-stranded molecule24 (left), a Möbius-type cycloparaphenylene with saturated linkers (–CH2O–) (ref. 34) (centre) and a Möbius-type sulfur-embedded belt35 (right). c, CNB, MCNB and their respective synthetic strategies.
Results and discussion
Here we report the synthesis, isolation and optical analysis of a Möbius carbon nanobelt (MCNB), that is, a fully fused CNB with a twist. The key to the synthesis of such MCNBs is a modification of our previously reported synthetic strategy for CNBs19,39. As shown in Fig. 1c, (n,n)CNBs (n = 6, 8 and 12, where (n,n) is the chiral index of the corresponding carbon nanotubes) were synthesized via a reductive homocoupling reaction using cyclic molecules that consisted of dibromoparaphenylene and cis-ethenylene precursors19,39. The important feature of this method is that a CNB can be generated when the number of repeat units is even, whereas an MCNB can be obtained when the number is odd. This is a simple but powerful method for the synthesis of a complex Möbius topology from highly symmetric precursors.
Strain energy calculation
The target size of the MCNB was determined using density functional theory (DFT) calculations. We found that MCNBs have a higher strain energy than CNBs of the same size (for details, see Supplementary Fig. 1), and that the strain of the MCNBs is mainly induced during the final bond-formation step. Figure 2a,b shows the hypothetical homodesmotic reactions using (n,n)MCNBs, (n,n)CNBs and their corresponding precursors (pre(n,n)MCNBs and pre(n,n)CNBs), based on which the strain induced in the final bond-formation step (ΔHFBF (kcal mol–1)) was estimated. Cis-stilbene and phenanthrene were used as reference molecules. For belts of a similar size, the ΔHFBF of the MCNB was much higher than that of the CNB (for example, (6,6)CNB, ΔHFBF = 40.2 kcal mol–1; (7,7)MCNB, ΔHFBF = 121.1 kcal mol–1). As (6,6)CNB was successfully synthesized using a nickel-mediated homocoupling reaction, the strain energy allowed by this synthetic method was estimated to be approximately 40 kcal mol–1. Based on these considerations, the synthetic pathway and the symmetry of the product, (15,15)MCNB (ΔHFBF = 51.1 kcal mol–1) and (25,25)MCNB (ΔHFBF = 29.6 kcal mol–1) were selected as the targets. The strain energies of the molecules were overall 85.7 and 49.4 kcal mol–1, respectively, which indicates that the strain decreases with increasing size of the MCNB (for details, see Supplementary Fig. 1).

a,b, Hypothetical homodesmotic reactions to estimate the strain increase during the final C–C-bond-formation steps for (n,n)MCNB (a) and (n,n)CNB (b). For pre(M)CNBs, the bromine atoms are replaced with hydrogen atoms for clarity. c, Plot of the heat of formation for the equations in a and b as a function of the size of the CNBs and MCNBs. For all the calculations, the B3LYP level of theory and 6-31G(d) basis sets were used.
Synthesis
Our synthetic route to the MCNBs is shown in Figs. 3 and 4. To improve the solubility of the intermediates and products, n-butoxy groups were introduced to the starting material 2. Thus, (25,25)MCNB with 20 butoxy groups (1) was targeted and synthesized from simple precursors 2 and 5 over 14 steps. First, the unsymmetric functionalization of phenanthrene 2 was investigated to ensure a Z-selective Wittig reaction sequence. During the screening of the Lewis-acid-catalysed formylation of 2, we found that monoformylated 3 was obtained selectively using TiCl4 and MeOCHCl2 in a high yield (75%), and that a subsequent chloromethylation with ZrCl4 and MeOCH2Cl smoothly afforded the bifunctional phenanthrene 4a with formyl and chloromethyl groups in an 84% yield. The formyl and chloromethyl groups of 4a were then converted into acetal and phosphonium groups, respectively, to yield 4b. The sequential Wittig reaction of 5 with 4a followed by 4b produced the key intermediate 7c (Fig. 3a). Starting from 7c as the monomer, its dimer (8c), trimer (9c) and pentamer (10c) were synthesized via Wittig reactions (Fig. 3b). In these reactions, the formyl and phosphonium groups reacted selectively, as the chloromethyl and dimethylacetal groups were inert under the reaction conditions. The macrocyclization was performed with 10d, which was derived from 10c and bore formyl and phosphonium groups to yield 11 in a 67% yield. The reductive coupling of 11 with Ni(cod)2 (cod, 1,8-cyclooctadiene) and 4,4′-methoxycarbonyl-2,2′-bipyridyl gave (BuO)20(25,25)MCNB (1) in a 20% yield (Fig. 4a). In contrast, only a trace mass peak corresponding to (BuO)12(15,15)MCNB for the macrocycle 12 was observed under similar conditions (Fig. 4b; see Supplementary Fig. 2 for details). According to DFT calculations, the failure to generate the (15,15)MCNB structure might be due to the huge strain required for the formation of the final bond (ΔHFBF = 51.1 kcal mol–1).

a, Synthesis of a repeating units 7a–7c. b, Preparation of precursor 10d by sequential Wittig reactions. Reagents and conditions: (i) TiCl4, MeOCHCl2, dichloromethane, –45 °C; (ii) ZrCl4, MeOCH2Cl, 1,2-dichloroethane, room temperature (r.t.); (iii) TsOH, CH(OMe)3, THF/methanol, r.t.; (iv) PPh3, CH(OMe)3, 80–90 °C; (v) DBU (1,8-diazabicyclo[5.4.0]undec-7-ene), dichloromethane, –10 °C; (vi) HCl(aq), THF, 40 °C; (vii) TMG (1,1,3,3-tetramethylguanidine), THF, r.t.; (viii) TFA (trifluoroacetic acid), chloroform, r.t.; (ix) LiHMDS (Li hexamethyldisilazide, molecular sieve 4A (MS4A), chloroform/THF, –78 °C to –60 °C. Ts, p-toluenesulfonyl.

a, Macrocyclization of 10d and Ni-mediated homocoupling to afford (BuO)20(25,25)MCNB (1). b, Unsuccessful attempt to synthesize (15,15)MCNB from cyclic precursor 12. Reagents and conditions: (i) iPr2NEt, MS4A, chloroform, 0 °C; (ii) Ni(cod)2, 4,4′-methoxycarbonyl-2,2′-bipyridyl, NMP (N-methylpyrrolidone), 70 °C. (iii) PPh3, CH(OMe)3, 90 °C; TFA, chloroform, r.t.; TMG, THF, r.t.
The thus obtained Möbius belt 1 was characterized using high-resolution mass spectrometry and NMR spectroscopy. The high-resolution mass spectrum showed an isotope pattern with its highest peak at 3,944.9449, which is in good agreement with the simulated pattern and mass number (m/z = 3,944.9423) expected for C280H260O20 (for details, see Supplementary Fig. 3). The DFT-optimized structure of 1 shows a C2-symmetry with a long (~38 Å) and a short (~30 Å) axis (Fig. 5a). The broadened aromatic signals in the 1H NMR spectrum observed at 25 °C converged at 140 °C into seven singlet signals, which can be assigned to a–h (shown in Fig. 5b) as supported by DFT calculations (see Supplementary Fig. 7 for details). These results indicate that the twist moiety of the Möbius belt moves quickly around the belt at a high temperature, as predicted for Möbius cyclacenes40. As shown in Fig. 5c, the molecular motion was simulated using a density functional tight binding with molecular dynamics (DFTB-MD) calculation (for details, see Materials and methods in the Supplementary Information).

a, The structure of 1 was optimized at the B3LYP/6-31G(d) level of theory, whereby the butoxy groups were replaced by methoxy groups. b, Aromatic region of the 1H NMR spectra of 1 in 1,1,2,2-tetrachloroethane-d2 at 25 °C and at 140 °C. c, Snapshots of the DFTB-MD simulation of (25,25)MCNB (carbon, red or blue; hydrogen, white). For the details, see Supplementary Video 1).
Photophysical properties
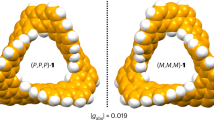
The photophysical properties of Möbius CNB 1 were also investigated. As shown in Fig. 6a, absorption maxima at 389 and 409 nm as well as a small absorption peak at 477 nm were observed, and greenish-blue fluorescence with maxima at 480, 513 and 551 nm were observed on excitation at 380 nm. Based on the fluorescence quantum yield (10%) and half-life (14.1 ns), the radiative and non-radiative decay rate constants (kr and knr) were estimated to be 7.1 × 106 and 6.4 × 107 s–1, respectively. Time-dependent DFT calculations of 1 suggested that, unlike in the D3h-symmetric (6,6)CNB, the S0 → S1 transition (assignable to the small band at 477 nm) is symmetry allowed (f = 0.6239), which reflects the lowered symmetry caused by the Möbius topology (Supplementary Fig. 4). The topological chirality of 1 was also examined experimentally. Chiral separation of 1 was successfully achieved using chiral HPLC, and the circular dichroism (CD) spectrum of each fraction was collected (Fig. 6b and Supplementary Figs. 5 and 6). Based on the CD spectra simulated using time-dependent DFT calculations (for details, see Supplementary Fig. 6), the first and second fractions were tentatively assigned to M and P chirality, respectively.

a, Absorption (solid line) and fluorescence (dashed line) of a dichloromethane solution of 1. Insets: photographs of a dichloromethane solution of 1 with (right) or without (left) ultraviolet light irradiation (254 nm). b, CD spectra of dichloromethane solutions of 1 separated using chiral column chromatography (CHIRALPAK-IE) with tentative structural assignments (M or P); butoxy groups are omitted for clarity. ε, absorption coefficient; τ, fluorescence lifetime; φ, fluorescence quantum yield.
Conclusion
In conclusion, we successfully synthesized a MCNB, that is, a topological molecular nanocarbon with a twist on armchair CNBs. The strategy of using a variant of the previously used CNB precursor, cyclo(dibromoparaphenylene-Z-ethenylene) with an odd number of units led to the discovery of a rational synthetic route to such MCNBs. DFT calculations of the intrinsic strain energies suggested that the synthesis of MCNBs with large sizes would be most promising, and therefore, (25,25)MCNB was selected as the target. The synthesis was carried out via Z-selective Wittig and intramolecular homocoupling reactions with nickel complexes to yield decabutoxylated (25,25)MCNB (1) over 14 steps. NMR spectroscopy and DFTB-MD calculations revealed that the Möbius twist structure moved quickly around the molecule in solution. Photophysical measurements revealed that the synthesized MCNB exhibited a greenish-blue fluorescence with a symmetry-allowed S0 → S1 transition caused by the lowered symmetry. Experimentally, chiral HPLC separation and CD spectroscopy revealed that the chirality originates from the Möbius topology. The combination of strain calculations with a rational synthetic strategy can be expected to create a variety of topological molecular nanocarbons, which will promote the progress of materials science in this area.
Methods
For the synthesis of (BuO)20(25,25)MCNB (1), to a 50 ml flask filled with argon gas were added Ni(cod)2 (130 mg, 0.473 mmol, 30 equiv.), 4,4‘-methoxycarbonyl-2,2′-bipyridyl (129 mg, 0.473 mmol, 30 equiv.) and NMP (9.6 ml). After the mixture was stirred at 70 °C for 1 h, the NMP (6.2 ml) solution of 11 (100 mg, 0.0157 mmol) was added and the resulting mixture was stirred at 70 °C for 20 min. The mixture was cooled to 0 °C and aqueous NH4Cl (12 ml) was added. The organic layer was extracted with chloroform, washed with brine, dried over Na2SO4 and then evaporated in vacuo. The crude product was purified by column chromatography (chloroform) and preparative thin-layer chromatography (hexane:chloroform:ethanol, 15:85:0.5) to afford (BuO)20(25,25)MCNB (1) (12.7 mg, 20%).
Data availability
Source data are provided with this paper. The experimental data and the characterization data for all of the compounds prepared in the course of these studies are provided in the Supplementary Information.
References
Kroto, H. W., Heath, J. R., O’Brien, S. C., Curl, R. F. & Smalley, R. E. C60: buckminsterfullerene. Nature 318, 162–163 (1985).
Iijima, S. Helical microtubules of graphitic carbon. Nature 354, 56–58 (1991).
Novoselov, K. S. et al. Electric field effect in atomically thin carbon films. Science 306, 666–669 (2004).
Yang, F. et al. Chirality pure carbon nanotubes: growth, sorting, and characterization. Chem. Rev. 120, 2693–2758 (2020).
Segawa, Y., Ito, H. & Itami, K. Structurally uniform and atomically precise carbon nanostructures. Nat. Rev. Mater. 1, 15002 (2016).
Segawa, Y., Levine, D. R. & Itami, K. Topologically unique molecular nanocarbons. Acc. Chem. Res. 52, 2760–2767 (2019).
Parekh, V. C. & Guha, P. C. Synthesis of pp′-diphenylenedimonosulphide. J. Indian Chem. Soc. 11, 95–100 (1934).
Tahara, K. & Tobe, Y. Molecular loops and belts. Chem. Rev. 106, 5274–5290 (2006).
Lewis, S. E. Cycloparaphenylenes and related nanohoops. Chem. Soc. Rev. 44, 2221–2304 (2015).
Segawa, Y., Yagi, A., Matsui, K. & Itami, K. Design and synthesis of carbon nanotube segments. Angew. Chem. Int. Ed. 55, 5136–5158 (2016).
Jasti, R., Bhattacharjee, J., Neaton, J. B. & Bertozzi, C. R. Synthesis, characterization, and theory of [9]-, [12]-, and [18]cycloparaphenylene: carbon nanohoop structures. J. Am. Chem. Soc. 130, 17646–17647 (2008).
Takaba, H., Omachi, H., Yamamoto, Y., Bouffard, J. & Itami, K. Selective synthesis of [12]cycloparaphenylene. Angew. Chem. Int. Ed. 48, 6112–6116 (2009).
Yamago, S., Watanabe, Y. & Iwamoto, T. Synthesis of [8]cycloparaphenylene from a square-shaped tetranuclear platinum complex. Angew. Chem. Int. Ed. 49, 757–759 (2010).
Matsui, K., Segawa, Y., Namikawa, T., Kamada, K. & Itami, K. Synthesis and properties of all-benzene carbon nanocages: a junction unit of branched carbon nanotubes. Chem. Sci. 4, 84–88 (2013).
Kayahara, E. et al. Synthesis and physical properties of a ball-like three-dimensional π-conjugated molecule. Nat. Commun. 4, 2694 (2013).
Matsui, K., Segawa, Y. & Itami, K. All-benzene carbon nanocages: size-selective synthesis, photophysical properties, and crystal structure. J. Am. Chem. Soc. 136, 16452–16458 (2014).
Hayase, N., Nogami, J., Shibata, Y. & Tanaka, K. Synthesis of a strained spherical carbon nanocage by regioselective alkyne cyclotrimerization. Angew. Chem. Int. Ed. 58, 9439–9442 (2019).
Segawa, Y. et al. Topological molecular nanocarbons: all-benzene catenane and trefoil knot. Science 365, 272–276 (2019).
Segawa, Y., Kuwayama, M. & Itami, K. Synthesis and structure of [9]cycloparaphenylene catenane: an all-benzene catenane consisting of small rings. Org. Lett. 22, 1067–1070 (2020).
Povie, G., Segawa, Y., Nishihara, T., Miyauchi, Y. & Itami, K. Synthesis of a carbon nanobelt. Science 356, 172–175 (2017).
Cheung, K. Y., Segawa, Y. & Itami, K. Synthetic strategies of carbon nanobelts and related belt-shaped polycyclic aromatic hydrocarbons. Chem. Eur. J. 26, 14791–14801 (2020).
Guo, Q.-H., Qiu, Y., Wang, M.-X. & Stoddart, J. F. Aromatic hydrocarbon belts. Nat. Chem. 13, 402–419 (2021).
Craik, D. J., Daly, N. L., Bond, T. & Waine, C. Plant cyclotides: a unique family of cyclic and knotted proteins that defines the cyclic cystine knot structural motif. J. Mol. Biol. 294, 1327–1336 (1999).
Walba, D. M., Richards, R. M. & Haltiwanger, R. C. Total synthesis of the first molecular Moebius strip. J. Am. Chem. Soc. 104, 3219–3221 (1982).
Ajami, D., Oeckler, O., Simon, A. & Herges, R. Synthesis of a Möbius aromatic hydrocarbon. Nature 426, 819–821 (2003).
Stępień, M., Latos-Grażyński, L., Sprutta, N., Chwalisz, P. & Szterenberg, L. Expanded porphyrin with a split personality: a Hückel–Möbius aromaticity switch. Angew. Chem. Int. Ed. 46, 7869–7873 (2007).
Sankar, J. et al. Unambiguous identification of Möbius aromaticity for meso-aryl-substituted [28]hexaphyrins(1.1.1.1.1.1). J. Am. Chem. Soc. 130, 13568–13579 (2008).
Yoon, Z. S., Osuka, A. & Kim, D. Möbius aromaticity and antiaromaticity in expanded porphyrins. Nat. Chem. 1, 113–122 (2009).
Schaller, G. R. et al. Design and synthesis of the first triply twisted Möbius annulene. Nat. Chem. 6, 608–613 (2014).
Naulet, G. et al. Cyclic tris-[5]helicenes with single and triple twisted Möbius topologies and Möbius aromaticity. Chem. Sci. 9, 8930–8936 (2018).
Fan, Y.-Y. et al. An isolable catenane consisting of two Möbius conjugated nanohoops. Nat. Commun. 9, 3037 (2018).
Jiang, X. et al. Kinetic control in the synthesis of a Möbius tris((ethynyl)[5]helicene) macrocycle using alkyne metathesis. J. Am. Chem. Soc. 142, 6493–6498 (2020).
Malinčík, J., Gaikwad, S., Boillat, M.-A., Häussinger, D. & Solomek, T. Figure-of-eight helicene carbon nanohoop with Möbius topology. Preprint at https://chemrxiv.org/engage/chemrxiv/article-details/60c754e30f50db4659397dda (2021).
Nishigaki, S. et al. Synthesis of belt- and Möbius-shaped cycloparaphenylenes by rhodium-catalyzed alkyne cyclotrimerization. J. Am. Chem. Soc. 141, 14955–14960 (2019).
Wang, S. et al. Sulphur-embedded hydrocarbon belts: synthesis, structure and redox chemistry of cyclothianthrenes. Angew. Chem. Int. Ed. 60, 18443–18447 (2021).
Herges, R. Topology in chemistry: designing Möbius molecules. Chem. Rev. 106, 4820–4842 (2006).
Türker, L. MNDO treatment of the Hückel and Möbius types of cyclacenes. J. Mol. Struct. Theochem 454, 83–86 (1998).
Türker, L. & Gümüş, S. Cyclacenes. J. Mol. Struct. Theochem 685, 1–33 (2004).
Povie, G., Segawa, Y., Nishihara, T., Miyauchi, Y. & Itami, K. Synthesis and size-dependent properties of [12], [16], and [24]carbon nanobelts. J. Am. Chem. Soc. 140, 10054–10059 (2018).
dos Santos, M. C. & Alvarez, F. Spin current in the Möbius cyclacene belts. Chem. Phys. Lett. 471, 276–279 (2009).
Acknowledgements
This work was supported by the ERATO program from JST (JPMJER1302 to K.I.), the Funding Program for KAKENHI from MEXT (JP1905463 to K.I.; JP19H02701 and JP19K22183 to Y.S.), a grant-in-aid for Scientific Research on Innovative Areas from the JSPS (‘π-Figuration’ JP17H05149 to Y.S. and ‘Coordination Asymmetry’ JP19H04570 to Y.H.), Toyoaki Scholarship Foundation (to Y.S.), Daiko Foundation (to Y.S.) and Asahi Glass Foundation (to Y.S.). K.W. acknowledges the Special Inter-University Researcher program in the Institute for Molecular Science. We thank K. Yonekura, K. Takaba, S. Maki-Yonekura, N. Yasuda, RIGAKU Co. and DAICEL Co. for support with the measurements. Calculations were partially performed using the Research Center for Computational Science(21-IMS-C177). The Institute of Transformative Bio-Molecules is supported by the World Premier International Research Center Initiative.
Author information
Authors and Affiliations
Contributions
K.I. and Y.S. conceived the project and prepared the manuscript with feedback from the other authors. T.W., K.Y., M.K. and K.W. performed the synthetic experiments and photophysical measurements. Y.S., K.W., J.P. and Y.H. performed the computational study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Synthesis thanks Jun Zhu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Alison Stoddart, in collaboration with the Nature Synthesis team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Experimental Section, Supplementary Fig. 1–7, Table 1, NMR spectra and refs. 1–14.
Supplementary Video 1
DFTB-MD simulation of (25,25)MCNB.
Supplementary Data 1
Cartesian coordinates of the optimized structures by using DFT calculations (xyz format).
Source data
Source Data Fig. 2
Plot of the heat of formation as a function of the size of the MCNB.
Source Data Fig. 4
Cartesian coordinates of the snapshots of the DFTB-MD simulation of (25,25)MCNB and 1H NMR spectra of 1 at 25 °C and 140 °C.
Source Data Fig. 5
Raw data of absorption, fluorescence and CD spectra of 1.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Segawa, Y., Watanabe, T., Yamanoue, K. et al. Synthesis of a Möbius carbon nanobelt. Nat. Synth 1, 535–541 (2022). https://doi.org/10.1038/s44160-022-00075-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s44160-022-00075-8
This article is cited by
-
Electronic and structural properties of Möbius boron-nitride and carbon nanobelts
Discover Nano (2024)
-
A catenane that is topologically achiral despite being composed of oriented rings
Nature Chemistry (2023)
-
Synthesis of branched silica nanotrees using a nanodroplet sequential fusion strategy
Nature Synthesis (2023)
-
Catalytic stereoselective synthesis of doubly, triply and quadruply twisted aromatic belts
Nature Synthesis (2023)
-
Paradromic molecules
Nature Synthesis (2023)
