
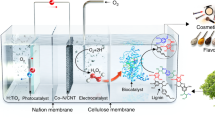
Abstract
Each year, the pulp and paper industry produces approximately 50 million metric tons of lignin as waste, 95% of which is combusted or abandoned. Here, we report the use of lignin as a photocatalyst that forms H2O2 by O2 reduction and H2O oxidation under visible light. We investigated the photophysical and electronic properties of two lignin models, lignosulfonate and kraft lignin, by spectroscopic and photoelectrochemical analyses, and demonstrated the photoredox chemistry of lignin using these and other lignin models (for example, native-like cellulolytic enzyme lignin, artificial lignin dehydrogenation polymer and phenolic β-aryl ether-type lignin dimer). Furthermore, the integration of lignin and H2O2-dependent unspecific peroxygenases (UPOs) enabled the highly enantioselective oxyfunctionalization of various C–H bonds. The use of lignin photocatalysts solves a number of the challenges relating to the sustainable activation of UPOs, notably, eliminating the need for artificial electron donors and suppressing the HO·-mediated inactivation of UPOs. Thus, the lignin–UPO hybrid catalyst achieved a total turnover number of UPO of 81,000 for solar-powered biocatalytic oxyfunctionalization in photochemical platforms.

This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others

Data availability
The data supporting the findings of this study are available within the article and its Supplementary Information. Source data are provided with this paper.
References
Kille, S., Zilly, F. E., Acevedo, J. P. & Reetz, M. T. Regio- and stereoselectivity of P450-catalysed hydroxylation of steroids controlled by laboratory evolution. Nat. Chem. 3, 738–743 (2011).
Gumulya, Y. et al. Engineering highly functional thermostable proteins using ancestral sequence reconstruction. Nat. Catal. 1, 878–888 (2018).
Zhang, W. et al. Selective aerobic oxidation reactions using a combination of photocatalytic water oxidation and enzymatic oxyfunctionalizations. Nat. Catal. 1, 55–62 (2018).
Lee, S. H., Choi, D. S., Kuk, S. K. & Park, C. B. Photobiocatalysis: activating redox enzymes by direct or indirect transfer of photoinduced electrons. Angew. Chem. Int. Ed. 57, 7958–7985 (2018).
Völler, J.-S. Enzymatic H2O2 for biocatalysis. Nat. Catal. 2, 375 (2019).
Zhang, W. et al. Selective activation of C−H bonds in a cascade process combining photochemistry and biocatalysis. Angew. Chem. Int. Ed. 56, 15451–15455 (2017).
Willot, S. J. P. et al. Expanding the spectrum of light-driven peroxygenase reactions. ACS Catal. 9, 890–894 (2019).
van Schie, M. M. C. H. et al. Cascading g-C3N4 and peroxygenases for selective oxyfunctionalization reactions. ACS Catal. 9, 7409–7417 (2019).
Kim, J. et al. Nicotinamide adenine dinucleotide as a photocatalyst. Sci. Adv. 5, eaax0501 (2019).
Choi, D. S., Kim, J., Hollmann, F. & Park, C. B. Solar-assisted eBiorefinery: photoelectrochemical pairing of oxyfunctionalization and hydrogenation reactions. Angew. Chem. Int. Ed. 59, 15886–15890 (2020).
Choi, D. S. et al. Bias-free in situ H2O2 generation in a photovoltaic-photoelectrochemical tandem cell for biocatalytic oxyfunctionalization. ACS Catal. 9, 10562–10566 (2019).
Kim, J. & Park, C. B. Shedding light on biocatalysis: photoelectrochemical platforms for solar-driven biotransformation. Curr. Opin. Chem. Biol. 49, 122–129 (2019).
Kim, J. et al. Robust FeOOH/BiVO4/Cu(In, Ga)Se2 tandem structure for solar-powered biocatalytic CO2 reduction. J. Mater. Chem. A 8, 8496–8502 (2020).
Kuk, S. K. et al. CO2-reductive, copper oxide-based photobiocathode for Z-scheme semi-artificial leaf structure. ChemSusChem 13, 2940–2944 (2020).
Lee, Y. W. et al. Unbiased biocatalytic solar-to-chemical conversion by FeOOH/BiVO4/perovskite tandem structure. Nat. Commun. 9, 4208 (2018).
Kim, J. et al. Biocatalytic C=C bond reduction through carbon nanodot-sensitized regeneration of NADH analogues. Angew. Chem. Int. Ed. 57, 13825–13828 (2018).
Le, T.-K. et al. Solar-powered whole-cell P450 catalytic platform for C-hydroxylation reactions. ChemSusChem 14, 3054–3058 (2021).
Son, G., Kim, J. & Park, C. B. Interference of solvatochromic twist in amyloid nanostructure for light-driven biocatalysis. ACS Appl. Energy Mater. 3, 1215–1221 (2020).
Wang, D., Lee, S. H., Kim, J. & Park, C. B. “Waste to wealth”: lignin as a renewable building block for energy harvesting/storage and environmental remediation. ChemSusChem 13, 2807–2827 (2020).
Chen, C.-C., Dai, L., Ma, L. & Guo, R.-T. Enzymatic degradation of plant biomass and synthetic polymers. Nat. Rev. Chem. 4, 114–126 (2020).
Li, C., Zhao, X., Wang, A., Huber, G. W. & Zhang, T. Catalytic transformation of lignin for the production of chemicals and fuels. Chem. Rev. 115, 11559–11624 (2015).
Wang, M. & Wang, F. Catalytic scissoring of lignin into aryl monomers. Adv. Mater. 31, 1901866 (2019).
Sanderson, K. Lignocellulose: a chewy problem. Nature 474, S12–S14 (2011).
Wang, D. et al. Lignin-fueled photoelectrochemical platform for light-driven redox biotransformation. Green Chem. 22, 5151–5160 (2020).
Romero, N. A. & Nicewicz, D. A. Organic photoredox catalysis. Chem. Rev. 116, 10075–10166 (2016).
Zhang, G., Lan, Z.-A. & Wang, X. Conjugated polymers: catalysts for photocatalytic hydrogen evolution. Angew. Chem. Int. Ed. 55, 15712–15727 (2016).
Dessbesell, L., Paleologou, M., Leitch, M., Pulkki, R. & Xu, C. Global lignin supply overview and kraft lignin potential as an alternative for petroleum-based polymers. Renew. Sustain. Energy Rev. 123, 109768 (2020).
Sáez-Jiménez, V. et al. Role of surface tryptophan for peroxidase oxidation of nonphenolic lignin. Biotechnol. Biofuels 9, 198 (2016).
Lancefield, C. S. et al. Identification of a diagnostic structural motif reveals a new reaction intermediate and condensation pathway in kraft lignin formation. Chem. Sci. 9, 6348–6360 (2018).
Crestini, C., Lange, H., Sette, M. & Argyropoulos, D. S. On the structure of softwood kraft lignin. Green Chem. 19, 4104–4121 (2017).
Constant, S. et al. New insights into the structure and composition of technical lignins: a comparative characterisation study. Green Chem. 18, 2651–2665 (2016).
Jiang, Z. et al. Nature-based catalyst for visible-light-driven photocatalytic CO2 reduction. Energ. Environ. Sci. 11, 2382–2389 (2018).
Perry, S. C. et al. Electrochemical synthesis of hydrogen peroxide from water and oxygen. Nat. Rev. Chem. 3, 442–458 (2019).
Kitahara, R., Yoshimura, Y., Xue, M., Kameda, T. & Mulder, F. A. A. Detecting O2 binding sites in protein cavities. Sci. Rep. 6, 20534 (2016).
Hou, H., Zeng, X. & Zhang, X. Production of hydrogen peroxide by photocatalytic processes. Angew. Chem. Int. Ed. 59, 17356–17376 (2020).
Yoon, J. et al. Piezobiocatalysis: ultrasound-driven enzymatic oxyfunctionalization of C–H bonds. ACS Catal. 10, 5236–5242 (2020).
Kim, J. K., Yang, J., Park, S. Y., Yu, J.-H. & Kim, K. H. Cellulase recycling in high-solids enzymatic hydrolysis of pretreated empty fruit bunches. Biotechnol. Biofuels 12, 138 (2019).
Hwang, H., Moon, S.-J., Won, K., Kim, Y. H. & Choi, J. W. Parameters affecting in vitro monolignol couplings during dehydrogenative polymerization in the presence of peroxidase and H2O2. J. Ind. Eng. Chem. 26, 390–395 (2015).
Kim, H. & Ralph, J. Solution-state 2D NMR of ball-milled plant cell wall gels in DMSO-d6/pyridine-d5. Org. Biomol. Chem. 8, 576–591 (2010).
Rahimi, A., Azarpira, A., Kim, H., Ralph, J. & Stahl, S. S. Chemoselective metal-free aerobic alcohol oxidation in lignin. J. Am. Chem. Soc. 135, 6415–6418 (2013).
Choi, D. S. et al. Photoelectroenzymatic oxyfunctionalization on flavin-hybridized carbon nanotube electrode platform. ACS Catal. 7, 1563–1567 (2017).
Simon, T. et al. Redox shuttle mechanism enhances photocatalytic H2 generation on Ni-decorated CdS nanorods. Nat. Mater. 13, 1013–1018 (2014).
Schultz, D. M. & Yoon, T. P. Solar synthesis: prospects in visible light photocatalysis. Science 343, 1239176 (2014).
Huang, C. et al. Unveiling the structural properties of lignin–carbohydrate complexes in bamboo residues and its functionality as antioxidants and immunostimulants. ACS Sustain. Chem. Eng. 6, 12522–12531 (2018).
Gomez de Santos, P. et al. Selective synthesis of the human drug metabolite 5′-hydroxypropranolol by an evolved self-sufficient peroxygenase. ACS Catal. 8, 4789–4799 (2018).
Zeng, Z. et al. A fluorescence-electrochemical study of carbon nanodots (CNDs) in bio- and photoelectronic applications and energy gap investigation. Phys. Chem. Chem. Phys. 19, 20101–20109 (2017).
Acknowledgements
This work was supported by the National Research Foundation (NRF; grant nos. NRF-2015R1A3A2066191 (C.B.P. and J.K.) and 2017M1A2A2087630 (Y.H.K.)) and the Global Ph.D. Fellowship Program (grant no. NRF-2019H1A2A1075810 (J.K.)), Republic of Korea. The authors thank G. Jung from B. Shin’s group (Korea Advanced Institute of Science and Technology) for permitting us to use a micro gas chromatograph.
Author information
Authors and Affiliations
Contributions
J.K. conceived/designed the research, performed the experiments, analysed the data and wrote the manuscript. C.B.P. supervised the research. T.V.T.N. and Y.H.K. provided lignin DHP, CEL and their HSQC spectra. F.H. supplied the UPOs. J.K., Y.H.K., F.H. and C.B.P. commented on the photo(bio)catalysis. J.K. and T.V.T.N. discussed the molecular structures of the lignins. All authors revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Synthesis thanks Han Sen Soo and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Thomas West was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Methods, Tables 1–8 and Figs. 1–33.
Source data
Source Data Fig. 4
Statistical source data.
Rights and permissions
About this article

Cite this article
Kim, J., Nguyen, T.V.T., Kim, Y.H. et al. Lignin as a multifunctional photocatalyst for solar-powered biocatalytic oxyfunctionalization of C–H bonds. Nat Synth 1, 217–226 (2022). https://doi.org/10.1038/s44160-022-00035-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s44160-022-00035-2
This article is cited by
-
Photocured room temperature phosphorescent materials from lignosulfonate
Nature Communications (2024)
-
Room-temperature phosphorescent materials derived from natural resources
Nature Reviews Chemistry (2023)
-
Visible light-exposed lignin facilitates cellulose solubilization by lytic polysaccharide monooxygenases
Nature Communications (2023)
-
Solar-powered biorefinery
Nature Synthesis (2022)
-
Photoelectrocatalytic biosynthesis fuelled by microplastics
Nature Synthesis (2022)
