
Abstract
Ecological interactions between marine bacteria and phytoplankton play a pivotal role in governing the ocean’s major biogeochemical cycles. Among these, members of the marine Roseobacter Group (MRG) can establish mutualistic relationships with phytoplankton that are, in part, maintained by exchanges of the organosulfur compound, dimethylsulfoniopropionate (DMSP). Yet most of what is known about these interactions has been derived from culture-based laboratory studies. To investigate temporal and spatial co-occurrence patterns between members of the MRG and DMSP-producing phytoplankton we analysed 16S and 18S rRNA gene amplicon sequence variants (ASVs) derived from 5 years of monthly samples from seven environmentally distinct Australian oceanographic time-series. The MRG and DMSP-producer communities often displayed contemporaneous seasonality, which was greater in subtropical and temperate environments compared to tropical environments. The relative abundance of both groups varied latitudinally, displaying a poleward increase, peaking (MRG at 33% of total bacteria, DMSP producers at 42% of eukaryotic phototrophs) during recurrent spring-summer phytoplankton blooms in the most temperate site (Maria Island, Tasmania). Network analysis identified 20,140 significant positive correlations between MRG ASVs and DMSP producers and revealed that MRGs exhibit significantly stronger correlations to high DMSP producers relative to other DMSP-degrading bacteria (Pelagibacter, SAR86 and Actinobacteria). By utilising the power of a continental network of oceanographic time-series, this study provides in situ confirmation of interactions found in laboratory studies and demonstrates that the ecological dynamics of an important group of marine bacteria are shaped by the production of an abundant and biogeochemically significant organosulfur compound.
Similar content being viewed by others
Introduction
Roseobacters are a globally ubiquitous group of heterotrophic bacteria found in marine surface waters [1]. In the open ocean, the marine Roseobacter Group (MRG) generally represents <8% of bacterial cells [2, 3], but in more productive coastal waters they are often dominant members of bacterial communities, particularly during phytoplankton blooms [4,5,6,7,8,9,10,11,12], and can comprise up to 25% of all bacteria [13, 14]. Roseobacters have been described as archetypical phytoplankton associates [15] and have been shown to be key players in the transformation of phytoplankton-derived dissolved organic matter (DOM), as well as potential mutualistic partners of some phytoplankton species [7, 16].
The ecological interactions between members of the MRG and phytoplankton have been shown to be underpinned by sometimes reciprocal exchanges of a diverse suite of organic molecules [7, 16]. Among these, organic sulfur compounds, including the phytoplankton secondary metabolite dimethylsulfoniopropionate (DMSP), are believed to be key currencies within MRG–phytoplankton interactions [17, 18]. DMSP is produced in large quantities by several marine phytoplankton species, with this single compound accounting for up to 10% of photosynthetically-derived carbon in the surface ocean [19, 20]. DMSP is consequently an important source of DOM for bacterial heterotrophy in the surface ocean, where it has been estimated to contribute up to 13% of carbon and 100% sulfur requirements of some marine bacteria [21, 22]. The MRG, like other abundant lineages of marine bacteria, including Pelagibacter (HTCC1062), can metabolise DMSP using multiple pathways. There are two primary degradation pathways for DMSP [23]: the DMSP lyase pathway, which cleaves DMSP to produce acrylate and the volatile gas, dimethylsulphide (DMS); and the DMSP demethylation pathway, which precludes DMS as a biproduct and results in the assimilation of DMSP-derived sulfur. The balance between these two pathways, which is largely determined by the composition and behaviour of the bacterial community, has profound biogeochemical implications [24]. This is because the ventilation of DMS (derived from the DMSP lyase pathway) from marine surface waters represents the largest source of biogenic sulfur in the atmosphere, where it can ultimately be converted to cloud condensation nuclei that increase albedo [25]. The distribution of the two DMSP degradation pathways among marine bacteria is not equal, as DMSP demethylation is more widespread than lyase [26], but the relative proportion of these genes can shift dependent on compositional changes in the bacterial community (including increases in members of the MRG) [27]. Furthermore, even within a given Roseobacter strain, there is evidence that the relative expression of these two genes can vary substantially according to different environmental conditions [28]. Cumulatively, changes in the relative occurrence and expression of these DMSP degradation pathways will potentially be a critical factor governing oceanic DMS production.
The important role of the MRG in marine sulfur cycling has been widely documented in laboratory-based studies, whereby isolates have been shown to grow from DMSP enrichment and subsequently produce DMS and acrylate [29], and have been reported to establish mutualistic associations with DMSP-producing phytoplankton [7, 16]. Members of the MRG have been shown to contain genes encoding DMSP demethylation (dmdA) and multiple lyase genes (dddD,L,P,W and Q) [30,31,32,33,34]. Evidence for the role of DMSP in MRG–phytoplankton interactions has also been inferred from short-term field-based studies that have reported MRGs as a dominant feature of bacterial communities during blooms of DMSP-producing phytoplankton [4, 10, 11, 35, 36].
The temporal and spatial distribution of the MRG has been very well characterised as a group, but recent focus on 16S rRNA gene sequences has shown that differences in biogeographic and seasonal patterns are evident when examining species or subclades of the MRG [37,38,39]. For instance, the CHAB-I-5 subclade occurs globally, from tropical to polar surface waters and represents a major component of the total bacteria community in the North Sea and Atlantic Ocean in summer [14, 40]. However, there is little evidence that this cluster is interacting with marine phytoplankton, i.e. there is no significant correlation between CHAB-I-5 and chlorophyll a and members are almost exclusively found in free-living fractions of bacterial communities [14]. Conversely, another MRG subclade, the Roseobacter clade affiliated (RCA) cluster, are notably absent in tropical and subtropical waters, but are among the most abundant bacterioplankton in temperate to (sub)polar environments, where their abundance is often positively correlated with chlorophyll a [13].
Emerging differences in biogeography, temporal dynamics and life-history traits within the MRG highlight the need to improve our understanding of the determinants of Roseobacter diversity and their links to phytoplankton, in particular high DMSP producers (e.g. Prorocentrum, Micromonas and Phaeocystis) and low DMSP producers (e.g. Thalassiosira, Skeletonema and Bathycoccus). Here we employed a substantial dataset of 16S rRNA and 18S rRNA gene sequences from the Australian Microbiome Initiative, collected across a unique continental-scale network of oceanographic time-series sites, to assess spatial and temporal patterns in the MRG and DMSP-producer communities. Our goal was to develop a more comprehensive environmental perspective on the ecology of a fundamentally important group of copiotrophic marine bacteria, their links to phytoplankton, and the marine sulfur cycle.
Methods
Sampling locations
To characterise patterns in the MRG relative abundance, diversity and associations with DMSP-producing phytoplankton, samples were collected from seven long-term oceanographic time-series sites situated in different regions of the Australian continental shelf (Supplementary Table S1). These National Reference Stations (NRS) span 30 degrees of latitude, with ship-board sampling conducted on a monthly or quarterly basis as part of the Australian Integrated Marine Observing System (IMOS). The Darwin NRS (12° 24.00 S, 130° 46.08 E) is located 9 km offshore from Darwin Harbour in the tropical north of Australia and experiences a monsoonal tropical climate. The Yongala NRS (19° 18.51 S, 147° 37.10 E) is located 22 km offshore and is located in the Great Barrier Reef (GBR) lagoon. This tropical site is also subject to monsoonal climate involving heavy rainfall events between December to April. The North Stradbroke Island NRS (27° 20.50 S, 153° 33.73 E) is located 12 km north-east of North Stradbroke Island (NSI), Queensland, and is influenced by the warm oligotrophic East Australian Current (EAC), with stratification of the water column and increased salinity between September to April [41]. The Port Hacking NRS (34° 05.00 S, 151° 15.00 E) is a long running time-series (since 1953) situated 7 km offshore, near the city of Sydney (population 4.3 million). This subtropical site is at times a transition zone between the cooler waters of the Tasman Sea and warm water intrusions of the EAC. The site can experience occasional upwellings via eddy and wind-driven currents that are linked with enhanced nutrient availability and phytoplankton biomass [42, 43]. The Maria Island NRS (42° 35.80 S, 148° 14.00 E) is located 9 km east of Maria Island, Tasmania. This temperate station is the southern-most NRS location and sits at the southern-most extent of the EAC, with evidence that longer EAC incursions are leading to rapid ocean warming in the region [41]. The water column at the Maria Island NRS is well-mixed year-round and is reported to have greater seasonal and inter-annual variation of microbial populations relative to subtropical sites on the east coast of Australia [44]. The Kangaroo Island NRS (35° 49.93 S, 136° 26.84 E) is located 10 km west of Kangaroo Island, South Australia. This subtropical site experiences periodic upwelling episodes in the austral winter [41]. The Rottnest Island NRS (32° 00.00 S, 115° 25.00 E) is located 4.5 km from Rottnest Island, which is 22 km off the western Australian mainland in the Indian Ocean. This site is heavily influenced by the Leeuwin Current, which transports warm water southward and as a result, sea-surface temperatures are up to 5 °C warmer at the NRS site compared to systems at similar latitudes (32° S).
Characterisation of physicochemical conditions
Water samples and accompanying physicochemical data were collected on a monthly basis at the Yongala, Port Hacking, North Stradbroke Island, Maria Island and Rottnest Island NRS, while quarterly sampling was conducted at Darwin and Kangaroo Island between February 2012 and August 2017. Physicochemical parameters including sea-surface temperature (°C) and salinity (PSU), chlorophyll a, dissolved oxygen (μmol L−1), and inorganic nutrients, nitrate/nitrite (μmol L−1), orthophosphate (μmol L−1), ammonia (μmol L−1), and silicate (μmol L−1) were measured in accordance with the IMOS NRS sampling protocol [45, 46] and were retrieved from the IMOS curated Australian Ocean Data Network Portal (https://portal.aodn.org.au/).
Sample collection and microbial DNA preparation
A detailed description of the marine microbial sample collection protocols within the Australian Microbiome Initiative is provided in Brown et al. [41]. Briefly, 2 L of seawater was collected from pre-determined depths (Supplementary Table S1) using Niskin bottles and filtered onto 0.2 μm pore Sterivex™ GP filter (Millipore, MA, USA). DNA was subsequently extracted from filters using a modified PowerWater® Sterivex™ DNA Isolation Kit (MOBIO laboratories, Carlsbad, CA, now Qiagen) [47].
Amplicon sequencing and bioinformatic analysis
Bacterial and eukaryotic assemblages were characterised using 16S rRNA and 18S rRNA gene sequencing, respectively. 16S and 18S rRNA amplicon PCR sequencing was performed using the Illumina MiSeq platform at the Ramaciotti Centre for Genomics at the University of New South Wales. The Australian Microbiome project, that commenced in 2011 utilises the V1–V3 hypervariable region of the prokaryotic 16S rRNA gene and differs from other more recent large-scale molecular projects (TARA Oceans Expedition, Malaspina Expedition and BIOGEOTRACES project) that use the V4–V5 region [48,49,50,51]. Direct comparisons between these primers are scarce, however, it has been reported that V1–V3 provided considerably higher estimates of SAR11 (an Alphaproteobacteria) abundance compared with estimates provided by V4–V5, although did not report any inconsistencies between Roseobacter diversity or abundance [52]. V1–V3 was amplified using the 27F (AGAGTTTGATCMTGGCTCAG) [53] and 519R (GWATTACCGCGGCKGCTG) primer pairing [54] under the following thermocycling conditions: 95 °C for 10 min; 35 cycles of 94 °C for 30 s, 55 °C for 10 s and 72 °C; followed by a final extension at 72 °C for 10 min. The V4 region of the 18S rRNA gene was amplified using the TAReuk454FWD1 (CCAGCASCYGCGGTAATTCC) and a modified TAReuk-Rev3 (ACTTTCGTTCTTGATYRATGA) primer [55], designed to be less discriminant against Haptophytes than the original TAReuk-Rev3 primer [56]. 18S rRNA was amplified under the following conditions: 98 °C for 30 s; 10 cycles of 98 °C for 10 s, 44 °C for 30 s and 72 °C for 15 s; 20 cycles of 98 °C for 10 s, 62 °C for 30 s and 72 °C followed by a final extension at 72 °C for 7 min. At the time of analysis, the Australian Microbiome Initiative (AMI) marine microbes database consisted of 1307 16S samples (Accession numbers are available in Supplementary Table S2) and 749 18S samples (Accession numbers are available in Supplementary Table S3) that were analysed using the methods detailed below. All sequences are now publicly available at the AMI data portal https://data.bioplatforms.com/organisation/about/australian-microbiome.
In order to derive the highest possible phylogenetic resolution, an unfiltered database of unique sequences was prepared. Briefly, the database was prepared by merging Illumina R1 and R2 reads using FLASH [57, 58]. Sequences with <3 reads per sequencing run were removed in addition to the removal of all sequences containing N’s. Sequences displaying an N at the terminal base were trimmed and retained within the dataset. All unique sequences were identified and mapped in each sample using -fastx_uniques from USEARCH v.10.0.240 [59, 60] to generate a sample by read abundance table. A total of 59,862 unique 16S rRNA sequences and 67,074 unique 18 S rRNA sequences were generated. Unique bacterial sequences were taxonomically classified using the SILVA v132 database with a 50% Bayesian probability cut-off [61, 62] and phytoplankton sequences were classified using the Protist Ribosomal Reference Database (PR2 version 12) [63]. The workflow to process unique sequences from the AMI database is downloadable from https://github.com/AusMicrobiome/amplicon/raw/1.0.0/docs/amplicon_analysis_workflow.docx and a comprehensive rationale for using unfiltered sequences can be found in Supplementary Methods.
To characterise MRG sequences, a library of 89 de-replicated representative 16S rRNA gene sequences derived from MRG genomes were compiled from www.roseobase.org. Previous genomic analyses of the MRG define the group as a paraphyletic subgroup of Rhodobacteraceae [64] and strains within the Roseobacter group have been identified to share 89% identity of 16S rRNA region [1, 65]. Therefore, all sequences annotated as Rhodobacterales were retrieved from our database. Identification of MRGs in our database were then achieved by clustering representative 16S sequences from Roseobase at 89% similarity with all Rhodobacterales sequences (434 bp, V1–V3) using the VSEARCH clustering tool with default parameters [66]. Across the entire dataset, 2222 16S amplicon sequence variants (ASVs) were clustered with known Roseobacters, identifying them as putative members of the MRG. Phylogenetic trees with putative MRG sequences, an outgroup sequence annotated as SAR11 clade 1a and known Roseobacters were assembled to visualise the MRG phylogeny (Supplementary Fig. S1). Phylogenetic trees were constructed using MEGA bioinformatic software (version 7) [67]. First MRG sequence alignments were generated using MUSCLE with default parameters [68] and the tree topology and confidence estimated using the Maximum-likelihood method (500 bootstraps, Tamura-Nei substitution model) [69, 70].
Identifying DMSP-producing phytoplankton in 18S rRNA sequences
Photosynthetic protists defined as 18S rRNA ASVs assigned to PR2 taxa: Chlorophyta, Dinophyta, Cryptophyta, Haptophyta, Ochrophyta, Cercozoa, Syndiniales and Sarcomonadea were extracted from the dataset, resulting in a subset of 10,875 18S ASVs. A curated bioinformatic pipeline was used to classify 18S ASVs as potential DMSP producers by incorporating previous measurements of cellular DMSP production in monocultures [58] to putatively assign the ability to produce DMSP to 18S ASVs based on phylogenetic inference [71]. First, full length 18S sequences of strains found to contain one (or more) DMSP synthesis gene (DSYB and TPMT1/TPMT2) (n = 164) or no DMSP synthesis gene (n = 180) were collected from the Marine Eukaryotic Transcriptome Sequencing Project (MMETSP) (Supplementary Table S4) [72] The 18S sequences were aligned with the eukaryotic small subunit ribosomal RNA Rfam (RF01960) using Infernal (v 1.1) [73] in order to build a reference phylogeny with RAxML (v 8.0) [74] using the GTRGAMMA model. A second alignment of the 10,875 unique 18S ASVs was created with Infernal and then pplacer [75] was used to place ASVs onto the reference phylogeny. 18S ASVs that had significant sequence similarity (posterior probability of 90%, likelihood < −4000) with one of the identified MMETSP strains with or without DMSP biosynthesis gene carriers were assumed to be DMSP producers or non-DMSP producers, respectively. A total of 3359 18S sequences were identified as likely DMSP-producing phytoplankton and 1053 18S sequences identified as likely non-DMSP producers (NoDP) (Supplementary Table S5). The DMSP-producing ASVs were further categorised as low DMSP producers (LoDP) or high DMSP producers (HiDP) based on SILVA taxonomic assignment (at Genus level) matching isolates with previously measured intracellular DMSP concentrations of less than, or greater than 50 mM DMSP, respectively [76]. Of the putatively identified DMSP-producing ASVs, 143 were related to 18S sequences of known HiDPs, and 74 were related to 18S sequences of known LoDPs. The pipeline identified 3142 ASVs as DMSP producers that did not belong to a genus of phytoplankton with previously characterised intracellular DMSP concentrations (herein referred to as UnDP). The remaining 18S ASVs (6,460) that were not classified from the pipeline were omitted from subsequent analyses.
Statistical analyses
To identify differences in the composition of the MRG across sites we performed multivariate Analysis of similarities (ANOSIM) with Bonferroni corrected post-hoc pairwise tests on the 50 most abundant MRG ASVs. To understand relationships between MRG ASVs and environmental parameters (salinity, temperature, ammonium, nitrate/nitrite, oxygen, phosphate, chlorophyll a and silicate) responsible for dissimilarity between sites, SIMPER analysis and Canonical Correspondence Analysis were performed on the 50 most abundant MRG ASV normalised abundances. To investigate seasonal and intra-annual variability of the community structure of the top 50 MRG and DMSP-producing phytoplankton the temporal distance between samples (time lag) was compared using Bray–Curtis (B–C) similarities and a Mantel correlogram of all sites by grouping month and year. Seasonal shifts in communities were statistically verified by pooling B–C similarities in 6-month sampling intervals (Austral Autumn-Winter; Mar, Apr, May, Jun, Jul, and Aug, and Austral Spring-Summer; Sep, Oct, Nov, Dec, Jan and Feb) and comparing the average similarity between 6 monthly intervals (e.g. Spring-Summer vs. Autumn-Winter) and twelve-monthly intervals (e.g. Spring-Summer vs. Spring-Summer) using a Kruskal–Wallis test (p < 0.05). To test for co-occurrence patterns between the 50 most abundant MRG and DMSP-producing phytoplankton in samples from all seven NRS stations over the years 2015–2017 (n = 749 samples), the maximal information-based non-parametric exploration (MINE) pipeline was used. MINE is a tool used to detect novel associations in large datasets and provides a suite of non-parametric exploration statistics that can be used to identify and characterise relationships in data [63]. As a point of reference, the 50 most abundant non-MRG bacterial ASVs were also included in the analysis. To minimise the chance of spurious correlations, only MINE results with a total information coefficient (TICe) value greater than zero (indicating presence of relationship) and a maximal information coefficient (MICe) value ≥0.178 (indicating a significant relationship, corresponding with a corrected p value < 0.001 for n = 749) were included [77]. All significant positive correlations (Spearman’s Rho > 0) were used to calculate the average correlation between MRGs/non-MRGs and HiDP, LoDP, and NoDP. Significant differences between the strength of positive correlations (Spearman’s Rho) between the top 50 MRG ASVs and top 50 non-MRG ASVs to HiDPs, LoDPs, and NoDPs were tested using a One-way ANOVA followed by Tukey-Kramer pairwise comparisons with Bonferroni corrections (q < 0.05). All non-homoscedastic (Levene’s test, p < 0.05) univariate statistical tests used Kruskal–Wallis test with Bonferroni corrected post-hoc comparisons (q < 0.05). An additional dataset (n = 385) limited by the number of samples that contained all of the desired contextual data (i.e. salinity, temperature, ammonium, nitrate/nitrite, oxygen, phosphate, chlorophyll a, and silicate) was included in MINE analysis to identify any significant correlations between MRGs and environmental variables. Strong correlations (adjusted p value < 0.001, Spearman’s Rho > 0.6) between MRGs and DMSP producers and MRGs and environmental variable were visualised using an Edge-weighted Force directed function in Cytoscape v3.71. All univariate tests were performed using SPSS version 17.0 (SPSS Statistics, Inc., Chicago, IL, USA) and multivariate tests were performed using PAST version 4.0 [78].
Results and discussion
Spatial and temporal patterns in MRG
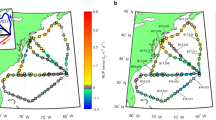
Our analysis of the overall relative abundance of the marine Roseobacter group (MRG) revealed continental-scale spatial patterns. The relative abundance of total MRG amplicon sequence variants (ASVs) was significantly greater at the subtropical and temperate sites of Port Hacking and Maria Island (Kruskal–Wallis, q < 0.05) compared to the other sites around Australia (Fig. 1). On average, MRG ASVs comprised 6.2 ± 0.34% and 5.7 ± 0.33% of the total bacterial community at Maria Island and Port Hacking respectively, whereas the lowest average relative abundance of MRG ASVs (1.8 ± 0.05%) was observed at the tropical site Yongala (Fig. 1). Overall, our results corroborate previous research showing that the MRG is widely distributed in marine environments and typically constitute 2–8% of surface water bacteria [2, 3]. The peak relative abundance of MRGs measured in samples at Maria Island (33.4% of total bacteria) and Port Hacking (22.4% of total bacteria) occurred above the thermocline (between 0 and 25 m depth) (Fig. 1). Patterns of increased MRG relative abundance in two of the sites on the south-eastern Australian coastal shelf (Port Hacking and Maria Island) might be explained by an overlap of environmental niches shared by dominant groups of the MRG, such as the CHAB-I-5 cluster [14, 40] and the Roseobacter clade affiliated cluster [13].

Total relative abundance (%) of the marine Roseobacter group (2222 unique 16S rRNA sequences) at the seven NRS between Jan′ 15 and Aug′ 17 at Darwin Harbour (DAR, Red), Yongala (YON, Orange), North Stradbroke Island (NSI, Yellow), Port Hacking Basin (PHB, Aqua), Maria Island (MAI, Dark Blue), Kangaroo Island (KAI, Cornflower Blue) and Rottnest Island (ROT, Green).
Temporal patterns in total MRG ASV relative abundance were also evident with significant differences found between months at all sites (Kruskal–Wallis, q < 0.05). Strong increases in total MRG relative abundance were measured in November to December at Port Hacking and between September to January at Maria Island, peaking in November 2015 and November 2016, respectively (Fig. 1). Notably, temporal increases in MRG relative abundance at Port Hacking and Maria Island are within the same range (as much as 20–40% of all 16S rRNA relative abundance) as MRG populations during spring-time phytoplankton blooms in other coastal locations [79, 80]. Exploration of the relationship between MRG relative abundance and phytoplankton biomass (chlorophyll a (chl a)) revealed significant positive correlations between these variables at Yongala, Rottnest Island, Kangaroo Island, Port Hacking and Maria Island (Supplementary Fig. S2). Furthermore, peaks of chl a at Port Hacking (16.9 mg chl a L−1) and Maria Island (1.6 mg chl a L−1) coincided with the MRG abundance comprising over 20% of the total bacterial abundance (Supplementary Fig. S2).
To further examine spatial and temporal patterns in the MRG we focused on a subset of the 50 relatively most abundant MRG ASVs across the entire dataset (Fig. 2A, Supplementary Table S6). These ASVs accounted for more than 75% of total MRG assigned 16S rRNA reads (2260 ASVs). All targeted ASVs were present at the temperate site, Maria Island, while only 19 of these ASVs occurred in the tropical site at Darwin (Fig. 2A). Significant dissimilarities in the composition of the targeted MRG communities were found between study sites (Fig. 2B, ANOSIM, R = 0.3758, p = 0.0001). The stations at Yongala, Kangaroo Island, and Maria Island each hosted unique MRG communities compared to all other sites (q < 0.05). The four remaining MRG assemblages at Darwin, Rottnest Island, North Stradbroke Island and Port Hacking did not differ from one another with exception of significantly different MRG communities at Port Hacking relative to Rottnest Island (q < 0.05). The latter pattern highlights differences between the Pacific Ocean and Indian Ocean bacterial communities, despite the sampling sites differing by only 2 degrees of latitude and both being characterised by subtropical conditions.

A Maximum-Likelihood phylogeny of 50 most abundant MRGs with known Roseobacter representative sequences (in bold). Presence (filled dot) or absence (unfilled dot) indicated at each time-series location. Coloured text corresponds with MRCs presented in panel B. Bootstrap values >70 are shown. B Canonical Correspondence Analysis (CCA) of top 50 MRG communities at DAR (Red), YON (Orange), NSI (Yellow), PHB (Aqua), MAI (Blue), KAI (Cornflower Blue) and ROT (Green). Vectors represent environmental parameters. The coloured boxes in CCA plot represent top 5 Roseobacters contributing to dissimilarity amongst MRG communities. Sequences of abundant MRG ASVs and known representatives in Supplementary Table S5.
Among environmental parameters, temperature, chlorophyll a, nitrate/nitrite and orthophosphate were variables correlated to MRG community dissimilarity (Fig. 2B). Temperature was most strongly correlated with MRG communities at Darwin, Yongala, North Stradbroke Island and Rottnest Island (Fig. 2B). Meanwhile, nutrients and chl a were correlated with MRG communities at Port Hacking, Kangaroo Island and Maria Island (Fig. 2B). Five abundant MRG ASVs collectively contributed to half of the average dissimilarity between sites (Fig. 2A, Supplementary Table S7). Biogeographical differences existed between the major contributors of dissimilarity (Fig. 2B). The bacterium (z2738) was the most abundant MRG across the entire dataset, representing an average of 43 ± 2.5% of the dominant MRG community (Fig. 3A). Rhodobacteraceae bacterium (z2738) was the dominant member of the MRG at subtropical (Rottnest Island and North Stradbroke Island) and tropical sites (Yongala and Darwin) that were correlated with temperature (Fig. 2B), along with Rhodobacteraceae bacterium (z4828) and HIMB11 (z2899) (Fig. 3A). Dominant members of the MRG at Port Hacking, Kangaroo Island and Maria Island were Amylibacter sp. (z3093) and Ascidiaceihabitans sp. (z2239) (Fig. 3A). Amylibacter sp. (z3093) was the most abundant ASV at Port Hacking and Maria Island, representing 20 ± 5.4% and 31 ± 5.5% of the dominant MRG community at the sites correlated to chl a and nutrients (phosphate and nitrate/nitrite) (Fig. 2B).

A Average monthly relative abundance of 50 most abundant MRG sequences in surface waters (<5 m depth) in the Australian Microbiome time-series between 2015 and 2017*. B Monthly relative abundance at Maria Island, Tasmania between Feb 2012 and Aug 2017. *2012–2017 for MAI, PHB and NSI.
In addition to biogeographic differences in the MRG community, temporal variability in the top 50 most abundant MRG ASVs was apparent at several of the sites, with levels of seasonality varying between sites (Fig. 3A). No seasonal variability in MRG community was evident in the tropical time-series at Darwin and Yongala (Fig. 4B, Supplementary Table S8). At these sites, similarities between sample pairs (time lags) ranged from 99.6 to 99.9% at Darwin and 75 to 80% at Yongala (Supplementary Fig. S3). On the other hand, seasonal changes in the MRG community, indicated by significantly lower similarity (q < 0.05) between communities at 6-month intervals relative to twelve-month intervals, occurred at the subtropical sites North Stradbroke Island, Rottnest Island and Port Hacking, and the temperate site at Maria Island (Fig. 4B, Supplementary Table S8). The greatest levels of seasonality were observed at Maria Island and Port Hacking, where intra-annual variability of MRG communities ranged from 25 to 60% and 27 to 48% similarity between sampling events, respectively (Fig. 4C). Seasonal differences at Port Hacking were punctuated by peaks in the relative abundance of Rhodobacteraceae bacterium (z2738), whereby this ASV represented 46.2% of the MRG community in autumn-winter, and peaks in the relative abundance of Amylibacter sp. (z3093), which made up almost 60% of the MRG community during spring-summer months (Fig. 3A). Similarly, at Maria Island, differences in the MRG community were also driven by Amylibacter sp. (z3093), which demonstrated significant increases in relative abundance (up to 62% of the community) during the Austral spring and summer months (Fig. 3B, ANOVA, F3 = 12.782, p < 0.05) and by Ascidiaceihabitans sp. (z2239), which displayed significant increases (up to 23% of the community) in relative abundance during the Austral summer (Fig. 3B, Kruskal–Wallis, q < 0.05). The strong seasonality in the dominant MRG community and significant positive correlations between total MRG relative abundance and chl a (Spearman’s Rho = 0.46, p < 0.01, n = 324, Supplementary Fig. S2) at Maria Island provides evidence that phytoplankton-derived DOM governs the dynamics of the MRG community in an environment known for predictable annual spring-summer phytoplankton blooms [45, 81].

A Australian National Reference Stations; Darwin (DAR, red), Yongala (YON, orange), North Stradbroke Island (NSI, yellow), Rottnest Island (ROT, green), Port Hacking (PHB, aqua), Kangaroo Island (KAI, cornflower blue) and Maria Island (MAI, dark blue). B Independent samples Kruskal–Wallis Test Statistic testing the null hypothesis of H0 = No difference exists in average Bray–Curtis Similarities between 6 monthly sampling and yearly sampling comparisons (Supplementary Table S8). Bars with asterisks (*) indicates that significant seasonal shifts are present. C Bray–Curtis similarities of top 50 MRG communities in all pairs of samples (y-axis) separated by different intervals of time (x-axis) in sites with significant seasonality.
Spatial and temporal patterns in DMSP-producing phytoplankton
To identify the occurrence of biogeographically and temporally conserved associations between MRGs and DMSP-producing phytoplankton, we characterised temporal and spatial patterns in DMSP producers and related these to MRG community dynamics. The spatial distribution of 3,350 ASVs identified as DMSP producers displayed similar continental-scale patterns as the MRG community, with greater total relative abundances (of the 18S phototrophic community) measured in subtropical and temperate waters than in tropical locations (Fig. 5). The greatest relative abundance of DMSP producers was seen at Maria Island, where DMSP-producing phytoplankton comprised an average of 15.2 ± 0.6% (at times representing up to 42%) of the 18S rRNA community (Fig. 5). The dominant DMSP-producer at tropical sites (Darwin and Yongala) was the high DMSP-producer (HiDP), Micromonas sp. (z27), which represented an average 2.6 ± 1.08% and 1.8 ± 2.32% of 18S rRNA sequences, respectively. At subtropical sites (North Stradbroke Island, Port Hacking and Rottnest Island), an increased relative abundance of the potential DMSP-producer Ostreococcus sp. (z8) (Unidentified DMSP-producer, UnDP, indicated by our pipeline, although no culture studies have confirmed this) was observed (Fig. 6, Kruskal–Wallis, q < 0.05). Meanwhile, significant increases in the relative abundance of the LoDP Bathycoccus sp. (z4) and the UnDP Ostreococcus sp. (z6) were found in more southerly sites (Kangaroo Island and Maria Island) compared to northern sites (Rottnest Island, Port Hacking, North Stradbroke Island, Yongala and Darwin) (Fig. 6, Kruskal–Wallis, q < 0.05). Dominant ASVs belonging to the HiDP genus Micromonas were also different across subtropical sites and temperate sites (Fig. 6), The relative abundance of Micromonas sp. (z10) was greatest at Rottnest Island (1.8 ± 1.69%, Kruskal–Wallis, q < 0.05), whereas Micromonas sp. (z22) (2.1 ± 1.75%, Kruskal–Wallis, q < 0.05) and Micromonas sp. (z221) (0.6 ± 0.89%, Fig. 6, Kruskal–Wallis, q < 0.05) were most abundant at Maria Island.

Total relative abundance (%) of DMSP-producing phytoplankton (3359 unique 18S rRNA sequences) in the Australian Microbiome Initiative Time-Series between Jan′ 15 and Aug′ 17 at locations, Darwin Harbour (DAR, Red), Yongala (YON, Orange), North Stradbroke Island (NSI, Yellow), Port Hacking Basin (PHB, Aqua), Maria Island (MAI, Dark Blue), Kangaroo Island (KAI, Cornflower Blue) and Rottnest Island (ROT, Green).

Average monthly relative abundance of 20 most abundant DMSP-producing phytoplankton (all depths) at Australian National Reference Stations; Darwin (DAR, red), Yongala (YON, orange), North Stradbroke Island (NSI, yellow), Rottnest Island (ROT, green), Port Hacking (PHB, aqua), Kangaroo Island (KAI, cornflower blue) and Maria Island (MAI, dark blue) between 2015 and 2017.
Temporal patterns in total DMSP-producer abundances were also clear at other locations, with highest levels observed at Rottnest Island and Maria Island during the spring-summer periods of 2015–2016 (Fig. 5). Annual peaks in relative abundance of identified DMSP producers at Rottnest Island were associated with a dominance of picoeukaryote ASVs; Micromonas sp. (z10) (7.2%), Bathycoccus sp. (z4) (2.8%) and Micromonas sp. (z27) (2.3%) (Fig. 6) in 2015 (August, 46 m depth), and Micromonas sp. (z10) (5.2%), Bathycoccus sp. (z4) (1.8%) and Micromonas sp. (z22) (1.3%) in 2016 (November, 46 m depth) (Fig. 6). Alternatively, temporal peaks in the relative abundance of the DMSP-producing community at Maria Island were dominated by picoeukaryotes and diatoms in 2015 (December 2015, 20 m depth), including Chaetoceros sp. (z42) (5.3%), Bathycoccus sp. (z4) (4.2%), Micromonas sp. (z221) (4.2%), Micromonas sp. (z22) (2.8%) and Micromonas sp. (z10) (2.3%) (Fig. 6). Whereas a peak in DMSP-producing ASVs at Maria Island in 2016 (September, 20 m depth) were dominated by a mixed community of picoeukaryote, prymnesiophyte and dinoflagellate organisms, specifically Heterocapsa sp. (z602) (2.8%), Micromonas sp. (z22) (2.8%) and Phaeocystis sp. (z1250) (1%) (Fig. 6).
Consistent trends between MRG and DMSP-producing phytoplankton were not limited to patterns in relative abundance, with greatest levels of seasonality in DMSP-producer diversity observed at the same locations as the largest seasonal shifts in the MRG community (Fig. 7B). Consistent with the patterns in MRG, there was no significant seasonality in DMSP-producer community composition at Darwin, Yongala and Kangaroo Island (Supplementary Fig. S4). Conversely, significant levels of seasonality were observed at Rottnest Island, North Stradbroke Island, Port Hacking and Maria Island (Fig. 7B, Supplementary Table S2).

A Australian National Reference Stations; Darwin (DAR, red), Yongala (YON, orange), North Stradbroke Island (NSI, yellow), Rottnest Island (ROT, green), Port Hacking (PHB, aqua), Kangaroo Island (KAI, cornflower blue) and Maria Island (MAI, dark blue). B Independent samples Kruskal–Wallis Test Statistic testing the null hypothesis of H0 = No difference exists in average Bray–Curtis Similarities between 6 monthly sampling and yearly sampling comparisons (Supplementary Table S9). Bars with asterisks (*) indicates significant seasonal shifts are present. C Bray–Curtis similarities of top 50 DMSP-producing ASVs in all pairs of samples (y-axis) separated by different intervals of time (x-axis) in sites with significant seasonality
Spatiotemporal coupling between Roseobacters and DMSP-producing phytoplankton
Our results indicate the existence of consistent spatial and temporal trends in abundance and levels of seasonality between communities of DMSP-producing phytoplankton and the MRG. Coherent patterns of seasonality between both groups of organisms occurred at several locations (Fig. 7). Moreover, a significant positive correlation (Spearman’s Rho = 0.28, p = 0.04, n = 749) between the total MRG relative abundance and DMSP-producer relative abundance (Spearman’s Rho = 0.28, p = 0.04, n = 749) further supports the hypothesis that members of the MRG are influenced by the potential availability of DMSP [15].
To more directly examine these potential ecological links, at the level of individual ASVs, we used a network analysis approach. Over 100,000 positive significant interactions were used to generate co-occurrence networks between DMSP-producing ASVs and heterotrophic bacteria. To test the hypothesis that DMSP-producing phytoplankton strongly influence the spatiotemporal dynamics of the MRG, we compared the strength and number of correlations between DMSP producers putatively classified by their potential cellular DMSP concentrations (high DMSP producers (HiDP), low DMSP producers (LoDP) and non-DMSP producers (NoDP) with the 50 most abundant MRGs. Overall, significantly greater average Spearman’s Rank correlations were present between MRGs and HiDPs compared to LoDPs and NoDPs (Fig. 8B). Additionally, no difference in average correlation was found between MRG relative abundance with LoDP or NoDP phytoplankton relative abundance (Fig. 8B). These differences suggest an important role HiDPs play in providing a preferential source of reduced sulfur to MRGs [82, 83]. Moreover, we reveal that LoDP and NoDP phytoplankton demonstrate a weaker impact on MRG abundance, most likely due to the release of lesser or no DMSP as a dissolved organic substrate. We also compared MRG–DMSP-producer correlations to other non-MRG bacterial lineages (e.g. Pelagibacterales, SAR86, Actinobacteria and Flavobacteriales) with DMSP producers. While there were not more correlations between MRG and DMSP producers, the correlations between MRG and HiDPs were significantly stronger than non-MRG lineages to HiDPs, LoDPs and NoDPs (Fig. 8B). This pattern is indicative of specific ecological associations between MRG and high DMSP-producing phytoplankton and is consistent with previous field studies that reported the dominance of Roseobacters in microbial communities associated with blooms of high DMSP-producing phytoplankton species including Emiliania huxleyi and Phaeocystis sp. and the isolation of Roseobacters from the high DMSP-producer Alexandrium sp. in laboratory experiments [10, 35, 84]. Our approach, which has examined a continental-scale dataset comprised of seven molecular time-series, spanning tropical to temperate environments of the Pacific and Indian Ocean, inclusive of two of the four major boundary currents of the Southern hemisphere provides a new and direct line of environmental evidence that Roseobacters are indeed key sulfur cycling lineages associated with prolific DMSP producers.

A Maximum-likelihood tree of top 50 MRGs and 46 most abundant other bacteria in the Australian-Microbiome time-series. Heatmap shows avg. Spearman’s Rho of all significant positive co-occurrences with high DMSP-producing phytoplankton (HiDP). B Comparison of avg. Spearman’s Rho between MRGs (pink) and bacteria (grey) with non DMSP-producers (NoDP), low DMSP producers (LoDP) and HiDP. Asterisks denotes significant difference as indicated by Tukey’s HSD (p < 0.05). Error bars are mean ± standard deviation.
A subset of strong significant positive interactions between the top 50 MRG and DMSP-producing phytoplankton was used to construct a network highlighting ASV-specific ecological interaction in our dataset (Fig. 9). A total of 22 of the 50 MRGs tested with phytoplankton ASVs had at least one significant positive interaction resulting in a Spearman’s Rho of greater than 0.6 (Fig. 9A). Of the phytoplankton ASVs (51 ASVs) with strong correlations to MRGs, ~18% were designated as HiDPs and 16% were LoDPs (Fig. 9A). Overall, 127 strong MRG–phytoplankton interactions (Spearman’s Rho > 0.6) were identified, consisting of 43 correlations to HiDPs and 26 to LoDPs (Fig. 9A, Supplementary Material S1). The strongest correlations across the entire network occurred between the MRG ASV identified as Sulfitobacter sp. (z3196) with the HiDP Phaeocystis sp. z1250 (Spearman’s Rho = 0.819) and LoDP Thalassiosira sp. (z1978) (Spearman’s Rho = 0.792) (Fig. 9B), providing evidence that Sulfitobacter sp. (z3196) represents a key associate of DMSP-producing phytoplankton. This Sulfitobacter sp. ASV was most prevalent at Kangaroo Island and Maria Island, where on average it represented 2.3% of the MRG community, and 0.2% of the total bacterial community at both sites (Fig. 3A). Notably, this ASV exhibited 5–10-fold increases in relative abundance in parallel to similar increases in relative abundances of Phaeocystis sp. z1250, which represented up to 2.4% of the eukaryotic community at Maria Island, and Thalassiosira sp. z1978, which represented up to 1.9% of the eukaryotic community at Kangaroo Island. This continental-scale co-co-occurrence between Sulfitobacter with DMSP producers, validates repeated identification and isolation of Sulfitobacter strains from DMSP-producing phytoplankton cultures [85,86,87] or blooms [88,89,90]. Moreover, the result is supported by recent experimental evidence describing the potential mechanism underpinning this co-occurrence, involving metabolic exchanges, including the transfer of DMSP, between Sulfitobacter (strain SA11) and the DMSP-producing diatom Pseudo-nitzschia multiseries that promoted growth of both partners [16]. The observed co-occurrence patterns across our seven oceanographic time-series are significant as they provide further in situ support for the existence of potentially symbiotic relationships between Sulfitobacter sp. and DMSP-producing phytoplankton [16].

A All Strong positive Roseobacter—DMSP-producer interactions. Coloured nodes show MRGs (as defined in Fig. 3) and grey nodes are eukaryotes. DMSP-producer type is designated by coloured halo to represent high DMSP-producers (HiDP, red), low DMSP-producers (LoDP, blue), unidentified DMSP-producers (UnDP, pink) and non DMSP-producers (NoDP, no halo). Size of nodes shows average relative abundance of 16S (Roseobacters) and 18S rRNA sequences (Phytoplankton) across all samples (n = 749). Nodes are edge-weighted force directed (Biolayout) based on strength of Spearman’s Rho. Thickness of edges represent Spearman’s Rho > 0.6 between an MRG—eukaryote. Environmental parameters shown as triangle nodes (Temp. temperature, NOx nitrate/nitrite). B Strongest interactions between Sulfitobacter and eukaryotes.
Several other dominant MRG ASVs displayed strong correlations to ASVs identified as the marine picoeukaryote Micromonas. Specifically, strong correlations involved Amylibacter sp. (z3093) and Micromonas sp. (z221) (Spearman’s Rho = 0.788), as well as Ascidiaceihabitans sp. (z2239) and Micromonas sp. (z22) (Spearman’s Rho = 0.748) and Rhodobacteraceae bacterium (z4828) with Micromonas sp. (z27) (Spearman’s Rho = 0.64) (Fig. 9A). Notably a laboratory isolate, Micromonas pusilla CCMP490 (NCBI accession AY955003.1) is a high DMSP-producing phytoplankton with measured intracellular concentrations as great as 161.9 mmol L−1 [76, 91], and is a similar organism (according to 18S rRNA gene sequences) to the ASVs Micromonas sp. (z22) (97% identity, 100% query cover), Micromonas sp. (z221) (97% identity, 100% query cover) and Micromonas sp. (z27) (96% identity, 100% query cover). Little is currently known about DMSP production by picoeukaryotes, as few isolates (n = 19 Chlorophyta and n = 2 Pelagophyta) have measured cellular DMSP content [58], though the recently reported methyltransferase enzymes required for DMSP synthesis have been detected in in situ picoeukaryote communities [92]. Additionally, previous modelling efforts have found that picoeukaryotes must be important DMSP producers, particularly in oligotrophic conditions, to account for DMSP production when other DMSP producers (e.g. diatoms, haptophytes) are not present/in bloom [76, 93]. Strong correlations between dominant MRG ASVs with abundant Micromonas ASVs in our results infer MRGs may display unexpected ecological links to, or even dependencies on, picoeukaryote derived DMSP.
Conclusion
Here we exploited the power of a continental-scale network of oceanographic time-series to provide a unique in situ assessment of the potential ecological links among marine Roseobacters and DMSP-producing phytoplankton. This approach revealed significant biogeographical and seasonal shifts in the composition of MRG that were often tightly coupled with repeating annual abundance patterns in eukaryotic phytoplankton likely responsible for producing DMSP. While we acknowledge the correlative nature of our results does not deliver irrefutable causal links between the two groups, we contend that large-scale and highly spatially and temporally resolved sampling efforts provide a powerful means of predicting community dynamics and inferring structure, particularly when a priori knowledge of the interaction mechanisms are available [94]. Using this approach, a number of novel and potentially significant ecological associations among often abundant members of the marine microbiome were identified, including new evidence for ecological associations between picoeukaryotes and specific members of the MRG. Our analysis is supported by two decades of laboratory and mesoscale interactions [4, 7, 10, 11, 35, 36, 80, 95,96,97] and delivers for the first time, ocean-scale field-based confirmation for the important ecological interactions between a key group of copiotrophic marine bacteria and DMSP-producing phytoplankton that are likely to play a pivotal role in carbon and sulfur cycling processes in the surface ocean.
References
Luo H, Moran MA. Evolutionary ecology of the marine Roseobacter clade. Microbiol Mol Biol Rev. 2014;78:573–87.
Wietz M, Gram L, Jørgensen B, Schramm A. Latitudinal patterns in the abundance of major marine bacterioplankton groups. Aquat Microbial Ecol. 2010;61:179–89.
Sunagawa S, Coelho LP, Chaffron S, Kultima JR, Labadie K, Salazar G, et al. Structure and function of the global ocean microbiome. Science. 2015;348:1261359.
González JM, Simó R, Massana R, Covert JS, Casamayor EO, Pedrós-Alió C, et al. Bacterial community structure associated with a dimethylsulfoniopropionate-producing North Atlantic algal bloom. Appli Environ Microbiol. 2000;66:4237–46.
González JM, Moran MA. Numerical dominance of a group of marine bacteria in the alpha-subclass of the class Proteobacteria in coastal seawater. Appl Environ Microbiol. 1997;63:4237–42.
Grossart HP, Levold F, Allgaier M, Simon M, Brinkhoff T. Marine diatom species harbour distinct bacterial communities. Environ Microbiol. 2005;7:860–73.
Amin SA, Parker MS, Armbrust EV. Interactions between diatoms and bacteria. Microbiol Mol Biol Rev. 2012;76:667–84.
Alavi M, Miller T, Erlandson K, Schneider R, Belas R. Bacterial community associated with Pfiesteria‐like dinoflagellate cultures. Environ Microbiol. 2001;3:380–96.
Jasti S, Sieracki ME, Poulton NJ, Giewat MW, Rooney-Varga JN. Phylogenetic diversity and specificity of bacteria closely associated with Alexandrium spp. and other phytoplankton. Appl Environ Microbiol. 2005;71:3483–94.
Zubkov MV, Fuchs BM, Archer SD, Kiene RP, Amann R, Burkill PH. Rapid turnover of dissolved DMS and DMSP by defined bacterioplankton communities in the stratified euphotic zone of the North Sea. Deep Sea Res Top Stud Oceanogr. 2002;49:3017–38.
Zubkov MV, Fuchs BM, Archer SD, Kiene RP, Amann R, Burkill PH. Linking the composition of bacterioplankton to rapid turnover of dissolved dimethylsulphoniopropionate in an algal bloom in the North Sea. Environ Microbiol. 2001;3:304–11.
Teeling H, Fuchs BM, Becher D, Klockow C, Gardebrecht A, Bennke CM, et al. Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science. 2012;336:608–11.
Voget S, Wemheuer B, Brinkhoff T, Vollmers J, Dietrich S, Giebel H-A, et al. Adaptation of an abundant Roseobacter RCA organism to pelagic systems revealed by genomic and transcriptomic analyses. ISME J. 2015;9:371–84.
Billerbeck S, Wemheuer B, Voget S, Poehlein A, Giebel H-A, Brinkhoff T, et al. Biogeography and environmental genomics of the Roseobacter-affiliated pelagic CHAB-I-5 lineage. Nat Microbiol. 2016;1:16063.
Buchan A, LeCleir GR, Gulvik CA, González JM. Master recyclers: features and functions of bacteria associated with phytoplankton blooms. Nat Rev Microbiol. 2014;12:686–98.
Amin S, Hmelo L, van Tol H, Durham B, Carlson L, Heal K, et al. Interaction and signalling between a cosmopolitan phytoplankton and associated bacteria. Nature. 2015;522:98–101.
Landa M, Burns AS, Durham BP, Esson K, Nowinski B, Sharma S, et al. Sulfur metabolites that facilitate oceanic phytoplankton–bacteria carbon flux. ISME J. 2019;13:2536–50.
Durham BP, Boysen AK, Carlson LT, Groussman RD, Heal KR, Cain KR, et al. Sulfonate-based networks between eukaryotic phytoplankton and heterotrophic bacteria in the surface ocean. Nat Microbiol. 2019;4:1706–15.
Howard EC, Henriksen JR, Buchan A, Reisch CR, Bürgmann H, Welsh R, et al. Bacterial taxa that limit sulfur flux from the ocean. Science. 2006;314:649–52.
Levine NM, Toole DA, Neeley A, Bates NR, Doney SC, Dacey JW. Revising upper-ocean sulfur dynamics near Bermuda: new lessons from 3 years of concentration and rate measurements. Environ Chem. 2016;13:302–13.
Kiene RP. Turnover of dissolved DMSP in estuarine and shelf waters of the northern Gulf of Mexico. In Biological and environmental chemistry of DMSP and related sulfonium compounds. Boston, MA: Springer; 1996. pp. 337–49.
Kiene RP, Linn LJ. Distribution and turnover of dissolved DMSP and its relationship with bacterial production and dimethylsulfide in the Gulf of Mexico. Limnol Oceanogr. 2000;45:849–61.
Curson AR, Todd JD, Sullivan MJ, Johnston AW. Catabolism of dimethylsulphoniopropionate: microorganisms, enzymes and genes. Nat Rev Microbiol. 2011;9:849–59.
Simó R. Production of atmospheric sulfur by oceanic plankton: biogeochemical, ecological and evolutionary links. Trends Ecol Evol. 2001;16:287–94.
Charlson RJ, Lovelock JE, Andreae MO, Warren SG. Oceanic phytoplankton, atmospheric sulphur, cloud albedo and climate. Nature. 1987;326:655–61.
Reisch CR, Moran MA, Whitman WB. Bacterial catabolism of dimethylsulfoniopropionate (DMSP). Front Microbiol. 2011;2:172.
Varaljay VA, Howard EC, Sun S, Moran MA. Deep sequencing of a dimethylsulfoniopropionate-degrading gene (dmdA) by using PCR primer pairs designed on the basis of marine metagenomic data. Appl Environ Microbiol. 2010;76:609–17.
Varaljay VA, Robidart J, Preston CM, Gifford SM, Durham BP, Burns AS, et al. Single-taxon field measurements of bacterial gene regulation controlling DMSP fate. ISME J. 2015;9:1677.
Ledyard KM, DeLong EF, Dacey JW. Characterization of a DMSP-degrading bacterial isolate from the Sargasso Sea. Arch Microbiol. 1993;160:312–8.
Todd JD, Kirkwood M, Newton-Payne S, Johnston AW. DddW, a third DMSP lyase in a model Roseobacter marine bacterium, Ruegeria pomeroyi DSS-3. ISME J. 2012;6:223–6.
Todd JD, Curson AR, Kirkwood M, Sullivan MJ, Green RT, Johnston AW. DddQ, a novel, cupin‐containing, dimethylsulfoniopropionate lyase in marine roseobacters and in uncultured marine bacteria. Environ Microbiol. 2011;13:427–38.
Todd J, Curson A, Dupont C, Nicholson P, Johnston A. The dddP gene, encoding a novel enzyme that converts dimethylsulfoniopropionate into dimethyl sulfide, is widespread in ocean metagenomes and marine bacteria and also occurs in some Ascomycete fungi. Environ Microbiol. 2009;11:1376–85.
Todd JD, Curson AR, Nikolaidou‐Katsaraidou N, Brearley CA, Watmough NJ, Chan Y, et al. Molecular dissection of bacterial acrylate catabolism–unexpected links with dimethylsulfoniopropionate catabolism and dimethyl sulfide production. Environ Microbiol. 2010;12:327–43.
Curson A, Rogers R, Todd J, Brearley C, Johnston A. Molecular genetic analysis of a dimethylsulfoniopropionate lyase that liberates the climate‐changing gas dimethylsulfide in several marine α‐proteobacteria and Rhodobacter sphaeroides. Environ Microbiol. 2008;10:757–67.
Delmont TO, Hammar KM, Ducklow HW, Yager PL, Post AF. Phaeocystis antarctica blooms strongly influence bacterial community structures in the Amundsen Sea polynya. Front Microbiol. 2014;5:646.
Stoica E, Herndl GJ. Bacterioplankton community composition in nearshore waters of the NW Black Sea during consecutive diatom and coccolithophorid blooms. Aquat Sci. 2007;69:413–8.
Giebel HA, Brinkhoff T, Zwisler W, Selje N, Simon M. Distribution of Roseobacter RCA and SAR11 lineages and distinct bacterial communities from the subtropics to the Southern Ocean. Environ Microbiol. 2009;11:2164–78.
Landa M, Blain S, Christaki U, Monchy S, Obernosterer I. Shifts in bacterial community composition associated with increased carbon cycling in a mosaic of phytoplankton blooms. ISME J. 2016;10:39–50.
Wemheuer B, Güllert S, Billerbeck S, Giebel H-A, Voget S, Simon M, et al. Impact of a phytoplankton bloom on the diversity of the active bacterial community in the southern North Sea as revealed by metatranscriptomic approaches. FEMS Microbiol Ecol. 2014;87:378–89.
Alonso-Gutiérrez J, Lekunberri I, Teira E, Gasol JM, Figueras A, Novoa B. Bacterioplankton composition of the coastal upwelling system of ‘Ría de Vigo’, NW Spain. FEMS Microbiol Ecol. 2009;70:493–505.
Brown MV, Van De Kamp J, Ostrowski M, Seymour JR, Ingleton T, Messer LF, et al. Systematic, continental scale temporal monitoring of marine pelagic microbiota by the Australian Marine Microbial Biodiversity Initiative. Sci Data. 2018;5:180130.
Ajani P, Hallegraeff G, Allen D, Coughlan A, Richardson A, Armand L, et al. Establishing baselines: a review of eighty years of phytoplankton diversity and biomass in southeastern Australia. Oceanogr Mar Biol. 2016;54:387–412.
Matear R, Chamberlain M, Sun C, Feng M. Climate change projection of the Tasman Sea from an eddy‐resolving ocean model. J Geophys Res Oceans. 2013;118:2961–76.
Ostrowski M, Seymour J, Messer L, Varkey D, Goosen K, Smith M, et al. Status of Australian marine microbial assemblages. In State and Trends of Australia’s Ocean Report, Integrated Marine Observing System (IMOS). 2020. https://doi.org/10.26198/5e16aa3e49e7f.
Lynch TP, Morello EB, Evans K, Richardson AJ, Rochester W, Steinberg CR, et al. IMOS National Reference Stations: a continental-wide physical, chemical and biological coastal observing system. PloS ONE. 2014;9:e113652.
Lynch T, Roughan M, Mclaughlan D, Hughes D, Cherry D, Critchley G, et al. A national reference station infrastructure for Australia - Using telemetry and central processing to report multi-disciplinary data streams for monitoring marine ecosystem response to climate change. In: OCEANS 2008. 2008. https://doi.org/10.1109/OCEANS.2008.5151856.
Appleyard SA, Abell G, Watson R. Tackling microbial related issues in cultured shellfish via integrated molecular and water chemistry approaches. Clayton: CSIRO Marine and Atmospheric Research; 2013.
Parada AE, Needham DM, Fuhrman JA. Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ Microbiol. 2016;18:1403–14.
Duarte CM. Seafaring in the 21st century: the Malaspina 2010 circumnavigation expedition. Limnol Oceanogr Bull. 2015;24:11–4.
Biller SJ, Berube PM, Dooley K, Williams M, Satinsky BM, Hackl T, et al. Marine microbial metagenomes sampled across space and time. Sci Data. 2018;5:1–7.
Logares R, Sunagawa S, Salazar G, Cornejo‐Castillo FM, Ferrera I, Sarmento H, et al. Metagenomic 16S rDNA Illumina tags are a powerful alternative to amplicon sequencing to explore diversity and structure of microbial communities. Environm Microbiol. 2014;16:2659–71.
Dadon-Pilosof A, Conley KR, Jacobi Y, Haber M, Lombard F, Sutherland KR, et al. Surface properties of SAR11 bacteria facilitate grazing avoidance. Nat Microbiol. 2017;2:1608–15.
Lane D. 16S/23S rRNA sequencing. In: Nucleic acid techniques in bacterial systematics. New York: John Wiley & Sons; 1991, pp. 115–75.
Lane DJ, Pace B, Olsen GJ, Stahl DA, Sogin ML, Pace NR. Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses. Proc Natl Acad Sci. 1985;82:6955–9.
Piredda R, Tomasino M, D’erchia A, Manzari C, Pesole G, Montresor M, et al. Diversity and temporal patterns of planktonic protist assemblages at a Mediterranean Long Term Ecological Research site. FEMS Microbiol Ecol. 2017;93.
Stoeck T, Bass D, Nebel M, Christen R, Jones MD, Breiner HW, et al. Multiple marker parallel tag environmental DNA sequencing reveals a highly complex eukaryotic community in marine anoxic water. Mol Ecol. 2010;19:21–31.
Andrews S. FastQC: a quality control tool for high throughput sequence data. Cambridge, United Kingdom: Babraham Bioinformatics, Babraham Institute; 2010.
Magoč T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–63.
Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–1.
Gordon A, Hannon G. Fastx-toolkit. FASTQ/A short-reads preprocessing tools. 2010;5. http://hannonlab.cshl.edu/fastx_toolkit/.
Yilmaz P, Parfrey LW, Yarza P, Gerken J, Pruesse E, Quast C, et al. The SILVA and “all-species living tree project (LTP)” taxonomic frameworks. Nucl Acids Res. 2014;42:D643–8.
Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–7.
Guillou L, Bachar D, Audic S, Bass D, Berney C, Bittner L, et al. The Protist Ribosomal Reference database (PR2): a catalog of unicellular eukaryote small sub-unit rRNA sequences with curated taxonomy. Nucl Acids Res. 2012;41:D597–604.
Simon M, Scheuner C, Meier-Kolthoff JP, Brinkhoff T, Wagner-Döbler I, Ulbrich M, et al. Phylogenomics of Rhodobacteraceae reveals evolutionary adaptation to marine and non-marine habitats. ISME J. 2017;11:1483–99.
Brinkhoff T, Giebel H-A, Simon M. Diversity, ecology, and genomics of the Roseobacter clade: a short overview. Arch Microbiol. 2008;189:531–9.
Rognes T, Flouri T, Nichols B, Quince C, Mahé F. VSEARCH: a versatile open source tool for metagenomics. PeerJ. 2016;4:e2584.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucl Acids Res. 2004;32:1792–7.
Felsenstein J. Evolutionary trees from gene frequencies and quantitative characters: finding maximum likelihood estimates. Evolution. 1981:1229–42.
Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol. 1993;10:512–26.
emcparland. emcparland/dmspOTUs: first release. 2021. https://doi.org/10.5281/zenodo.5090864.
Keeling PJ, Burki F, Wilcox HM, Allam B, Allen EE, Amaral-Zettler LA, et al. The Marine Microbial Eukaryote Transcriptome Sequencing Project (MMETSP): illuminating the functional diversity of eukaryotic life in the oceans through transcriptome sequencing. PLoS Biol. 2014;12:e1001889.
Nawrocki EP, Eddy SR. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 2013;29:2933–5.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3.
Matsen FA, Kodner RB, Armbrust EV. pplacer: linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinform. 2010;11:538.
McParland EL, Levine NM. The role of differential DMSP production and community composition in predicting variability of global surface DMSP concentrations. Limnol Oceanogr. 2019;64:757–73.
Reshef DN, Reshef YA, Sabeti PC, Mitzenmacher M. An empirical study of the maximal and total information coefficients and leading measures of dependence. Ann Appl Stat. 2018;12:123–55.
Hammer Ø, Harper DA, Ryan PD. PAST: paleontological statistics software package for education and data analysis. Palaeontol Electron. 2001;4:9.
Buchan A, González JM, Moran MA. Overview of the marine Roseobacter lineage. Appl Environ Microbiol. 2005;71:5665–77.
Moran MA, González JM, Kiene RP. Linking a bacterial taxon to sulfur cycling in the sea: studies of the marine Roseobacter group. Geomicrobiol J. 2003;20:375–88.
Harris G, Nilsson C, Clementson L, Thomas D. The water masses of the east coast of Tasmania: seasonal and interannual variability and the influence on phytoplankton biomass and productivity. Mar Freshw Res. 1987;38:569–90.
Kiene RP, Linn LJ, González J, Moran MA, Bruton JA. Dimethylsulfoniopropionate and methanethiol are important precursors of methionine and protein-sulfur in marine bacterioplankton. Appl Environ Microbiol. 1999;65:4549–58.
Raina J-B, Tapiolas D, Willis BL, Bourne DG. Coral-associated bacteria and their role in the biogeochemical cycling of sulfur. Appl Environ Microbiol. 2009;75:3492–501.
Brinkmeyer R, Rappé M, Gallacher S, Medlin L. Development of clade-(Roseobacter and Alteromonas) and taxon-specific oligonucleotide probes to study interactions between toxic dinoflagellates and their associated bacteria. Eur J Phycol. 2000;35:315–29.
Töpel M, Pinder MI, Johansson ON, Kourtchenko O, Clarke AK, Godhe A. Complete genome sequence of novel Sulfitobacter pseudonitzschiae Strain SMR1, isolated from a culture of the marine diatom Skeletonema marinoi. J Genomics. 2019;7:7.
Hong Z, Lai Q, Luo Q, Jiang S, Zhu R, Liang J, et al. Sulfitobacter pseudonitzschiae sp. nov., isolated from the toxic marine diatom Pseudo-nitzschia multiseries. Int J Syst Evol Microbiol. 2015;65:95–100.
Yang Q, Ge Y-M, Iqbal NM, Yang X, Zhang X-l. Sulfitobacter alexandrii sp. nov., a new microalgae growth-promoting bacterium with exopolysaccharides bioflocculanting potential isolated from marine phycosphere. Antonie Van Leeuwenhoek. 2021;114:1091–106.
Ankrah NY, Lane T, Budinoff CR, Hadden MK, Buchan A. Draft genome sequence of Sulfitobacter sp. CB2047, a member of the Roseobacter clade of marine bacteria, isolated from an emiliania huxleyi bloom. Genome Announc. 2014;2:e01125–14.
Kwak M-J, Lee J-S, Lee KC, Kim KK, Eom MK, Kim BK, et al. Sulfitobacter geojensis sp. nov., Sulfitobacter noctilucae sp. nov., and Sulfitobacternoctilucicola sp. nov., isolated from coastal seawater. Int J Syst Evol Microbiol. 2014;64:3760–7.
Zhang F, Fan Y, Zhang D, Chen S, Bai X, Ma X, et al. Effect and mechanism of the algicidal bacterium Sulfitobacter porphyrae ZFX1 on the mitigation of harmful algal blooms caused by Prorocentrum donghaiense. Environ Pollut. 2020;263:114475.
Keller MD. Dimethyl sulfide production and marine phytoplankton: the importance of species composition and cell size. Biol Oceanogr. 1989;6:375–82.
McParland EL, Lee MD, Webb EA, Alexander H, Levine NM. DMSP synthesis genes distinguish two types of DMSP producer phenotypes. Environ Microbiol. 2021;23:1656–69.
Galí M, Simó R. A meta‐analysis of oceanic DMS and DMSP cycling processes: disentangling the summer paradox. Glob Biogeochem Cycles. 2015;29:496–515.
Carr A, Diener C, Baliga NS, Gibbons SM. Use and abuse of correlation analyses in microbial ecology. ISME J. 2019;13:2647–55.
Amin S, Hmelo L, Van Tol H, Durham B, Carlson L, Heal K, et al. Interaction and signalling between a cosmopolitan phytoplankton and associated bacteria. Nature. 2015;522:98.
Miller TR, Hnilicka K, Dziedzic A, Desplats P, Belas R. Chemotaxis of Silicibacter sp. strain TM1040 toward dinoflagellate products. Appl Environ Microbiol. 2004;70:4692–701.
Seymour JR, Simó R, Ahmed T, Stocker R. Chemoattraction to dimethylsulfoniopropionate throughout the marine microbial food web. Science. 2010;329:342–5.
Acknowledgements
This research was supported by Australian Research Council grants FT130100218 and DP180100838 awarded to JRS and DP140101045 awarded to JRS and KP, as well as an Australian Government Research Training Programme Scholarship awarded to JO. We also gratefully acknowledge the Australian Microbiome Initiative and the Integrated Marine Observing System (IMOS) for the generation of data used in this publication. The Australian Microbiome initiative is supported by funding from Bioplatforms Australia and IMOS through the Australian Government’s National Collaborative Research Infrastructure Strategy (NCRIS), Parks Australia through the Bush Blitz programme funded by the Australian Government and BHP, and the Commonwealth Scientific and Industrial Research Organisation (CSIRO). Oceanographic data was sourced from IMOS, which is enabled by NCRIS.
Author information
Authors and Affiliations
Contributions
JO, JRS and KP designed the project. MO and ARB processed the sequences data. JO analysed the dataset. ELM and NML developed the DMSP-producer pipeline used in the study. Data was curated by JO, ARB, MO and JVDK. Funding was acquired by JRS and KP. Writing, review and editing of the paper was performed by JO, AB, MO, MB, JVDK, LB, LFM, KP, ELM, NML, NS, KP and JRS.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
O’Brien, J., McParland, E.L., Bramucci, A.R. et al. Biogeographical and seasonal dynamics of the marine Roseobacter community and ecological links to DMSP-producing phytoplankton. ISME COMMUN. 2, 16 (2022). https://doi.org/10.1038/s43705-022-00099-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43705-022-00099-3

{kind=link}