
Abstract
Aging is typified by a progressive decline in mitochondrial activity and stress resilience. Here, we review how mitochondrial stress pathways have pleiotropic effects on cellular and systemic homeostasis, which can comprise protective or detrimental responses during aging. We describe recent evidence arguing that defects in these conserved adaptive pathways contribute to aging and age-related diseases. Signaling pathways regulating the mitochondrial unfolded protein response, mitochondrial membrane dynamics, and mitophagy are discussed, emphasizing how their failure contributes to heteroplasmy and de-regulation of key metabolites. Our current understanding of how these processes are controlled and interconnected explains how mitochondria can widely impact fundamental aspects of aging.
Similar content being viewed by others

Main
Mitochondria are evolutionarily derived from alphaproteobacteria that evolved in symbiosis within eukaryotic cells1. Although most alphaproteobacterial genes were transferred to the eukaryotic nucleus, mitochondria retained their genome to translate the remaining protein-coding genes within their DNA. This requires complex coordination of the transcription and translation from two genomes, and the import and processing of proteins into the mitochondria in an ever-changing cellular milieu2,3. Disruption of these finely controlled processes has been shown to impair cellular homeostasis. To cope with this downside of endosymbiosis, mitochondria have evolved multiple stress-response pathways. The mitochondrial stress-response (MSR) network contributes to the reconstitution of cellular homeostasis by preventing mitochondrial proteotoxicity and by redistributing and removing irreversibly damaged elements of the mitochondria4. In recent years, we have gained considerable insights into why a decline in the robustness of these MSR pathways contributes to cellular damage and organismal deterioration. This is underlined by our emerging understanding of how different types of mitochondrial defects are co-regulated and interact across cellular and systemic processes.
Here, we describe the pleiotropic effects of mitochondrial dysfunction in aging. We outline the major mitochondrial stress pathways, how their failure is interconnected with the expansion of mitochondrial DNA mutations and deregulated metabolism, and how this affects cellular and organismal homeostasis. We furthermore provide an integrated map of how combined mitochondrial defects impact several features of aging, suggesting conserved links that could potentially be harnessed to slow the aging process. We refer readers to other comprehensive reviews on topics not covered in depth here, such as cellular senescence, stem cell function, and reactive oxygen species (ROS)5,6,7.
Mitochondrial stress responses in aging and longevity
Mitochondrial unfolded protein response
Appropriate handling and folding of proteins are essential, especially in mitochondria, whose proteome is encoded in both the nuclear and mitochondrial genomes. Mitochondrial protein homeostasis is ensured by an elaborate protein quality-control network composed of molecular chaperones and proteases8 governed by the mitochondrial unfolded protein response (UPRmt) (Fig. 1). Upon mitochondrial proteotoxic stress, the UPRmt induces the expression of chaperones, proteases, and other stress-response genes, mediated by activating transcriptional factor associated with stress-1 (ATFS-1) in Caenorhabditis elegans, and activating transcription factor 4 (ATF4) along with ATF5 and DNA damage inducible transcript 3 (DDIT3, also known as CHOP) in mammals, to restore mitochondrial function and adapt to stress9,10,11,12. Several conditions that interfere with mitochondrial proteostasis, such as an increased load of unassembled, damaged, or unfolded proteins, play a substantial part in UPRmt activation, with important implications in aging and longevity.

Stressors that induce proteotoxicity in the mitochondria such as accumulation of unassembled, unfolded, and damaged proteins can trigger the UPRmt. The activation of the UPRmt in C. elegans involves the digestion of unfolded or unassembled mitochondrial proteins by the mitochondrial matrix protease CLPP-1 and the transport of the fragmented peptides to the cytoplasm by HAF-1. Cytosolic accumulation of these mitochondrial peptides is at the heart of UPRmt activation. ATFS-1 is a transcription factor that, under basal conditions, localizes to the mitochondria, where it is constantly degraded by the Lon protease 1 (LONP-1). During mitochondrial stress, mitochondrial protein import is limited by the cytosolic accumulation of mitochondrial peptides. ATFS-1 then can shuttle to the nucleus12, where, in cooperation with other co-factors including DVE-1 and UBL-5, as well as epigenetic modulators including JMJDs and CBP-1 (refs. 247,248), it orchestrates the expression of a broad set of genes involved in mitochondrial quality control and metabolism12,252,253,254,255. In mammals, several types of mitochondrial perturbations (such as oxidative or proteotoxic stress), trigger the ISR through the phosphorylation of translation initiation factor eIF2α, which shuts down global translation and favors the cap-independent translation of the ATF4 transcription factor, leading to the expression of UPRmt and cytoprotective genes9,49. In addition to ATF4, at least two other transcription factors, ATF5 and CHOP, and several epigenetic modulators247,248 are involved in UPRmt activation10,11. The attenuation of general protein translation, together with the transcriptional induction of proteostasis genes, such as those encoding chaperones and proteases, thus antagonize cellular proteotoxicity.
Disruption of most of the electron transport chain (ETC) subunits extends the lifespan in C. elegans, yeast, flies, and mice13,14,15,16,17,18. It is well established that activation of the MSR is a critical component of mitochondrial-stress-induced longevity. In worms, knockdown of oxidative phosphorylation (OXPHOS) complexes I, III, IV, and V, all encoded in both mitochondrial DNA (mtDNA) and nuclear DNA (nDNA), triggers the UPRmt and extends lifespan18, while disruption of complex II, which is encoded only by nDNA, does not affect longevity19. Consistent with these results, complex-IV-deficient mice also show activation of the UPRmt and have a prolonged lifespan14,20. These data suggest that a mismatch between mtDNA- and nDNA-encoded ETC subunits, resulting in unassembled ETC components and the subsequent mitonuclear protein imbalance, is sufficient to drive the UPRmt and lifespan extension in worms and mammals. In agreement, the reduced expression of Mrps5, which encodes a mitochondrial ribosomal protein that regulates the translation of mtDNA-encoded ETC genes, induces a mitonuclear imbalance resulting in activation of the UPRmt, which correlates with an increased lifespan in the BXD mouse genetic reference population (GRP)21. In C. elegans, mrps-5 RNA interference (RNAi) increased the lifespan by more than 50%, highlighting an evolutionarily conserved mechanism linking the UPRmt to longevity21. In addition, pharmacologically inhibiting mitochondrial translation by using antibiotics that inhibit bacterial, and, hence, mitochondrial translation, such as doxycycline or chloramphenicol, induces the UPRmt and extends the health span and lifespan across kingdoms of life, from animals (C. elegans)21 to plants (Arabidopsis thaliana)22,23. Interestingly, as shown in yeast, worms, and mammals, such adaptive changes in mitochondrial translation can also affect cytosolic translation24,25,26, suggesting that cross-compartment synchronization is essential to maintain protein homeostasis during mitochondrial stress.
Mitonuclear protein imbalance also contributes to the lifespan extension seen upon mitochondrial biogenesis. An increased protein-folding workload in the mitochondria can be perceived as proteostatic stress, resulting in activation of the UPRmt (ref. 21). This has been demonstrated by the lifespan-extending effect of resveratrol, a well-known inducer of mitochondrial biogenesis, which also triggers the UPRmt in worms21. Likewise, genetic or pharmacological restoration of nicotinamide adenine dinucleotide (NAD+) in C. elegans induces mitochondrial biogenesis and promotes longevity via induction of mitonuclear imbalance and UPRmt (ref. 27). These findings translate to mammals, as restoring NAD+ levels by nicotinamide riboside (NR) administration in mice aged 24 months induces the UPRmt and extends the lifespan28. However, when treatment with NAD+ boosters was started in mice at 8 months of age, they were reported not to increase the lifespan29.
There are specific spatiotemporal requirements for UPRmt activation and longevity. In C. elegans, cco-1 and mrps-5 RNAi21,30 and RNAi targeting other respiratory chain components18, as well as doxycycline treatment, result in activation of the UPRmt and promote longevity only when the perturbation occurs early in life, before the L3/L4 larval stage. This suggests the existence of a surveillance system that monitors mitochondrial activity early in life and establishes the rate of the aging process throughout adulthood through epigenetic modulation (Box 1). Moreover, the longevity effect of UPRmt activation has been shown to be tissue-specific, as ETC disruption in neurons and intestine, but not in muscle, increases longevity in an UPRmt-dependent manner30. UPRmt activation by mitochondrial stress can also signal in a cell-nonautonomous manner to inform distant tissues of emanating mitochondrial stress. In worms, ETC inhibition or expression of toxic polyglutamine (polyQ) protein in neurons activates the UPRmt in the intestine, suggesting the existence of extracellular signals that inform the whole organism of stress and prepare against it30,31,32. Accordingly, the Wnt/EGL-20 ligand of the Frizzled receptor is a signaling molecule secreted by neurons upon mitochondrial stress, which triggers the UPRmt in peripheral tissues in the same organism32 and across generations, conferring stress resistance and longevity in the descendants33. Interestingly, this transgenerational stress-protective inheritance by Wnt signaling is caused by increased mtDNA in the germline leading to a mitonuclear protein imbalance and UPRmt (ref. 33). Likewise, the increased lifespan observed in Drosophila upon mild mitochondrial disruption requires inter-organ cross-talk involving UPRmt and insulin-like growth factor-binding protein 7, which systemically antagonizes insulin signaling34. In mice, growth differentiation factor 15 (GDF15) and fibroblast growth factor 21 (FGF21) were identified as UPRmt-associated hormones promoting metabolic benefits, such as improved insulin sensitivity and protection against hepatic steatosis35,36 as well as increased lifespan37,38. As these molecules are secreted during mitochondrial stress, they have often been termed ‘mitokines’; however, as they are not directly released by mitochondria, the term ‘metabokines’ may be more appropriate.
In contrast to mild and acute (time-restricted) mitochondrial stress, which improves organismal homeostasis, chronic OXPHOS dysfunction is often detrimental. For example, several mouse models with mitochondrial defects have a reduced lifespan39,40,41, and most of the human diseases associated with OXPHOS dysfunction are typified by protracted defects and hence are deleterious42,43. Circulating levels of FGF21 and GDF15 are substantially increased in mouse models of mitochondrial dysfunction44,45 and in human mitochondrial disorders45,46 and aging47, which might represent an attempt of the organism to cope with the sustained stress. Moreover, the UPRmt is, on the one hand, strongly induced in worm and mouse models of mtDNA deletion44,48,49, whereas, on the other hand, it seems to be required for the propagation of mtDNA deletion in worms48. Collectively, these results suggest that prolonged activation of mitochondrial stress can be harmful, whereas precise, mild mitochondrial stress exerts a beneficial adaptive effect on organismal aging. This mitohormetic effect of the UPRmt is in line with its temporal specificity, in which the UPRmt needs to be inflicted in the larval stages to extend worm lifespan.
The UPRmt pathway also modulates stem cell function in aging. In muscle stem cells of aged mice, restoring NAD+ levels with NR activates the SIRT1-dependent UPRmt, improving mitochondrial metabolism and attenuating senescence linked to increased lifespan28. Similarly, increasing NAD+ levels by overexpressing NAMPT, the rate-limiting NAD+ salvage enzyme, ameliorates cell senescence in aged mesenchymal stem cells50. The effects of NAD+-induced UPRmt signaling have also been shown to benefit muscular dystrophies, as it not only prevents MuSC senescence28, but also attenuates skeletal muscle and heart deterioration in mouse models of muscle dystrophy51. Also, SIRT7, which is downregulated upon aging and controls the expression of several mitochondrial proteins in mice52, improves the regenerative capacity of aged hematopoietic stem cells through a mechanism involving the activation of the UPRmt (ref. 53). Thus, the UPRmt may be essential to maintain stem cell function during aging by preventing senescence.
Defects in how mitochondria sense and respond to stress are crucial for initiating organismal decline in aging. The UPRmt emerged as an essential regulator of aging and longevity by synchronizing mitochondrial and nuclear genomes at the proteome level to maintain proper mitochondrial function upon stress. Suggestive of potential relevance in mammals, the UPRmt network is active in mouse and human populations across multiple tissues54 and is deregulated in several human age-related diseases, such as sarcopenia55 and Alzheimer’s disease56,57.
Mitochondrial membrane dynamics
Mitochondria are dynamic organelles; they can be found as isolated organelles, fused in large networks, and even unequally distributed in the cytosol by organized mitochondrial transport and positioning58. Mitochondrial fusion and fission, termed mitochondrial membrane dynamics, are essential components of the MSR. Through dilution and segregation of damaged organelles, cells ensure homeostasis and survival upon stress59,60. Thus, cellular and organismal health relies on tight regulation of mitochondrial fission and fusion, and disruption in any of these pathways is linked to aging and several age-related diseases59.
In mammals, fusion of the mitochondria is mediated by the GTPases mitofusin 1 (MFN1) and MFN2, which merge the outer mitochondrial membrane (OMM), and by optic atrophy 1 (OPA1), responsible for merging the inner mitochondrial membrane (IMM). Mitochondrial fission is predominantly orchestrated by the dynamin-related protein 1 (DRP1) GTPase in coordination with other OMM-associated receptors, such as mitochondrial fission factor (MFF) and mitochondrial fission 1 protein (FIS1)61,62. Both fission and fusion seem to facilitate the segregation and removal of dysfunctional mitochondria63. Moreover, DRP1 mediates two distinct types of mitochondrial fission: division at the midzone results in mitochondrial proliferation, whereas division in the periphery enables damaged material to be destined for mitophagy64 (Fig. 2a).

a, In mammals, fusion of mitochondria is regulated by two mitofusins (MFN1 and MFN2), proteins of the dynamin-related family of large GTPases, located in the OMM, and OPA1, located at the IMM. Fusion initiates with the docking of two MFN1 proteins in the mitochondria, inducing conformational changes that drive GTP hydrolysis with subsequent fusion of the mitochondrial OMMs. At the IMM, unprocessed OPA1, known as long OPA1 (L-OPA1), is cleaved by the peptidases OMA1 and YME1L to form the short form of OPA1 (S-OPA1), which, in association with cardiolipin, facilitates the fusion of the adjacent IMM following OPA1-dependent GTP hydrolysis. Fission is predominantly orchestrated by the dynamin-related protein 1 (DRP1). When dephosphorylated by calcineurin at Ser637, DRP1 translocates from the cytosol to the mitochondrial surface. There, DRP1 binds to its OMM receptors mitochondrial fission factor (MFF), mitochondrial dynamics protein of 49 kDa (MID49), MID51, and mitochondrial fission 1 protein (FIS1). DRP1 then oligomerizes and induces GTP-hydrolysis-membrane constriction. DRP1 also mediates mitochondrial peripheral fission, enabling damaged material to be destined for mitophagy. b, PINK1, a mitochondrial serine/threonine-protein kinase, senses impaired mitochondria and signals to the cytosolic E3 ligase parkin. Under basal conditions, PINK1 is imported to the mitochondria by TOM and TIM translocases, leading to the proteolytic cleavage of PINK1 by mitochondrial proteases. Upon stress, the IMM depolarizes and inhibits protein import. Uncleaved PINK1 hence accumulates in the OMM and activates parkin through direct phosphorylation of the parkin Ub-like (UBL) domain or through the phosphorylation of ubiquitin. Activated parkin additionally ubiquitinates multiple substrates in the OMM to recruit autophagy receptors, including p62, optineurin (OPTN), and NDP52, which facilitate the recruitment of LC3 and engulfment of impaired mitochondria by autophagosomes. Ubiquitin-independent mitophagy is regulated by the recruitment of autophagy receptors, such as BNIP3, NIX, and FUNDC1, to the mitochondrial membrane. These receptor proteins then recruit LC3, enabling the engulfment of mitochondria by the autophagosomes.
The link between mitochondrial fission and fusion in aging was initially observed in lower organisms. In two fungal aging models, reducing mitochondrial fission by Dnm1p (homolog of mammalian drp-1) deletion enhances lifespan65. In worms, fragmentation of the mitochondrial network and swollen mitochondria are observed with aging21,66,67, and diverse longevity pathways are associated with increased mitochondrial fusion68. Accordingly, inhibition of mitochondrial fusion abrogates the lifespan extension in long-lived mutant worms66,68. Aging manifests itself differently in distinct tissues, as in the case of germline cells, which connect generations and are essentially immortal. In worms, these cells avoid transmitting damage to the next generation by an invigoration of proteostasis, requiring a switch from fragmented to an elongated, fused mitochondrial network69.
However, mitochondrial fusion is not always analogous to lifespan extension. In Drosophila, reduction in mitofusion levels caused by overexpression of parkin, an E3 ubiquitin ligase involved in the ubiquitin–proteasome system and mitophagy, attenuates mitochondrial fusion and triggers mitochondrial fission, leading to an increase in multiple markers of mitochondrial activity and lifespan extension70. Alternatively, triggering mitochondrial fission through upregulation of Drp1 in the midlife of flies preserves mitochondrial respiratory function and prolongs lifespan71. Moreover, reducing mitochondrial translation by mrps-5 RNAi in C. elegans extends the lifespan while triggering mitochondrial fragmentation21. In this setting, altering fission or fusion synergizes with reduced mitochondrial translation to prolong the worm’s lifespan72. Simultaneous ablation of both mitochondrial fission and fusion produces opposing phenotypes in yeast and C. elegans. In yeast, this double ablation shortens the lifespan73, whereas in C. elegans, it extends the lifespan by increasing the homeostasis, fatty acid oxidation, and peroxisomal function in the mitochondrial network74.
In mice, both fission and fusion are impaired with age. Aged mice demonstrate reduced DRP1 activity and alterations in mitochondrial morphology in several tissues, including skeletal muscle, neurons, and oocytes75,76. Interestingly, both muscle-specific DRP1 overexpression or DRP1 knockdown in 18-month-old mice causes muscle atrophy77. Recently, it has been shown that the RNA-binding protein pumilio 2 (PUM2) increases with age in worms, mice, and humans67. PUM2 prevents Mff translation, suggesting a potential mechanism by which mitochondrial fission is impaired in aging67. Finally, ablation of both DRP1-mediated fission and MFN-mediated fusion in mice accelerates mitochondrial senescence in the heart78. These findings suggest that mitochondrial fission and fusion critically contribute to aging when not properly balanced.
Mitochondrial fusion is essential for maintaining mtDNA stability by diluting mtDNA mutations. Expansion of mtDNA mutations has been linked to age-associated mitochondrial decline in several species (see ‘mtDNA integrity in aging’). This notion was extensively studied using proofreading-deficient POLG mutator mice. This mouse model was engineered to contain proofreading-deficient mitochondrial DNA polymerase (POLG) through substitution of an alanine residue for aspartate on the POLG catalytic subunit (p.D257A), resulting in accumulation of mutated mtDNA and consequently accelerated aging39,40. Interestingly, while the mutator mouse survives into adulthood, crossing this strain with knocked out Mfn1 results in mitochondrial dysfunction and embryonic lethality79. Of note, people with OPA1 mutations present mtDNA instability as indicated by multiple mtDNA deletions80,81. These findings suggest that mitochondrial fusion is essential for diluting mutated mtDNA. However, it cannot be ruled out that the effect of membrane dynamics on mtDNA propagation may have tissue and temporal specificity. Indeed, fragmentation of the mitochondria is essential for removing mutant mtDNA in germline tissues of Drosophila, providing evidence for a fission-based selection against deleterious mtDNA mutations82, which seems opposite to the beneficial effects of fusion in the C. elegans germline69.
Mitophagy
When mitochondrial stress accumulates to levels that exceed the capacity of stress responses, autophagy of the mitochondria, termed mitophagy, takes place. Among all mitochondrial quality-control systems, mitophagy is the only one that mediates the turnover of the whole organelle, thus avoiding cellular damage and apoptosis. In higher eukaryotes, mitophagy operates in different cell types and tissues through ubiquitin-dependent and ubiquitin-independent pathways (Fig. 2b). The ubiquitin-dependent mechanism is mediated by the PINK1–parkin axis, which ubiquitinates multiple substrates to recruit autophagy receptors. Ubiquitin-independent mitophagy is regulated by direct recruitment of autophagy receptors, such as BNIP3, NIX, and FUNDC1. Both pathways culminate in the engulfment of mitochondria by autophagosomes83.
There is accumulating evidence that mitophagy affects aging and the lifespan in different organisms84. In C. elegans, mitophagy is required for lifespan extension of several long-lived mutants, including worms with reduced insulin–IGF-1 signaling or impaired mitochondrial function and mutants subjected to caloric restriction85. Furthermore, moderate mitochondrial deficiency and hypoxia response promote longevity in worms in a mitophagy-dependent manner86. In line with these results, deficiency in dct-1 and bec-1, both key autophagy genes, recapitulates the effect of aging on mitochondrial mass in young adult worms85. In Drosophila, parkin null mutants exhibit reduced lifespan and locomotor defects driven by muscle degeneration87, whereas parkin overexpression reduces proteotoxicity and extends lifespan70. Consistent with these findings, the longevity effect of mitochondrial perturbation in flies involves systemic repression of insulin signaling, facilitating mitophagy by enhancing lysosome biogenesis34.
Mitophagy decline has been observed in several tissues in mice upon aging. In a transgenic mouse strain expressing the fluorescent mitophagy reporter mt-Keima, a decrease of ~70% in mitophagy was observed in the hippocampus of 21-month-old mice compared with that in young 3-month-old mice88. On a similar note, loss of parkin in POLG mutator mice causes a massive loss of dopaminergic neurons by 1 year of age, suggesting that Parkin prevents neuronal deterioration following mitochondrial mutagenesis89. Mitophagy also declines in the mouse heart upon aging, contributing to OXPHOS dysfunction and heart failure90. Moreover, defective mitophagy was observed in muscle-specific stem cells isolated from aged mice and humans91. This was associated with mitochondrial dysfunction and senescence, which can be restored by re-establishing mitophagy91. In accordance with these findings, boosting mitophagy improves OXPHOS function in aged worms and mice67,92 and in a muscular-dystrophy mouse model, typified by accelerated muscle degradation93. Like in lower organisms, hypoxia also promotes mitophagy in mammals94, in which it has been shown to protect against mitochondrial toxicity and extend the lifespan in a genetic mouse model of mitochondrial disease95. Consistent with these results, hypoxic preconditioning attenuates ischemia and reperfusion injury through mitophagy in mice96. Mitophagy thus might represent a conserved strategy to maintain mitochondrial quality in hypoxic conditions.
A variety of mitophagy modulators have been shown to mitigate the effects of aging. Urolithin A (UA), a gut-microbiome-derived natural compound, induces mitophagy both in vitro and in vivo following oral administration in mice and humans92,97. In C. elegans, UA prevents the accumulation of dysfunctional mitochondria with age and extends lifespan92. These effects translate to rodents: UA improved muscle health in two mouse models of age-related muscle decline92 and a mouse model of muscular dystrophy, resulting in an increased survival rate93. Furthermore, UA treatment lowered protein aggregation and prevented cognitive impairment in animal models of Alzheimer’s disease98. The positive effects on mitochondrial health upon oral consumption of UA and its favorable safety profile97 have recently been documented in humans99. Other classes of compounds, such as actinonin, spermidine, and NAD+ enhancers, also exert their beneficial effects in models of aging and age-related disease through the enhancement of mitophagy56,98,100,101,102,103.
Mitophagy in inflammaging
Defective mitophagy has emerged as a central contributor to inflammation, which may underlie the age-dependent increase in low-grade inflammation, termed inflammaging. Parkin-deficient mice challenged with immunogenic stressors, such as low-dose lipopolysaccharide (LPS), develop Parkinson’s-disease-like symptoms, including loss of dopaminergic neurons and motor defects104. These phenotypes were also observed in aged Parkin–/– mutator mice105 and Pink1–/– mice infected with Gram-negative bacteria106. Interestingly, PINK1 and parkin were shown to repress mitochondrial antigen presentation delivered by mitochondrial-derived vesicles, thus suppressing an immune response provoked by inflammation107.
Independently of pathogen infection, immune responses can be triggered by intracellular molecules from senescent or dying cells, termed damage-associated molecular patterns (DAMPs)108,109. mtDNA release is a potent DAMP, activating both intracellular and extracellular immune pathways. When released in the cytosol, mtDNA stimulates the NLRP3 inflammasome, resulting in IL-1β and IL-18 secretion and apoptosis. Additionally, cells can sense mtDNA in the cytosol through the cyclic GMP–AMP synthase (cGAS), which is activated by double-stranded DNA, leading to the production of 2′3′ cyclic GMP–AMP (cGAMP), a second messenger molecule and agonist of the stimulator of interferon genes (STING). Mitophagy can prevent inflammation by promoting mtDNA clearance from damaged mitochondria, thus preventing cytosolic mtDNA release and subsequent STING1 activation105. Consistent with these data, mitophagy restrains inflammasome activation in macrophages by reducing cytosolic accumulation of mtDNA110,111. Furthermore, mitophagy flux is involved in the inflammatory responses mediated by IRGM1, a master regulator of type I interferon112, and can attenuate inflammation by directly restraining NLRP3-inflammasome overactivation in macrophages111. These findings support a role for mitophagy in restraining innate immune pathways. However, in acute inflammatory conditions, such as sepsis, mitophagy can exert opposite effects. For example, in a mouse model of sepsis caused by polymicrobial infection, pharmacological inhibition of mitophagy promotes macrophage activation favoring bactericidal clearance, leading to a higher survival rate113. Accordingly, mitochondrial modulators that promote mitophagy led to immunoparalysis, a secondary immune suppression in sepsis, counteracting the removal of infectious agents and worsening survival113.
Circulating mtDNA increases gradually with age and correlates with serum inflammatory markers114, suggesting a causal role of extracellular mtDNA in age-related innate immune activation. One proposed explanation is that age-related failure of mitochondrial quality control could increase the release of mitochondrial-derived DAMPs; for instance, mitochondrial fusion seems to regulate TLR9-mediated NF-κB activation in skeletal muscle of mice through mtDNA115. However, recent findings have challenged this notion, showing that a large portion of cell-free mtDNA in the blood is contained within whole mitochondria and does not circulate as naked DNA116. Additionally, cell-free mtDNA varies in response to common physiological stressors, such as exercise and psychological stress, suggesting that not all forms of cell-free mtDNA are pro-inflammatory117.
Integration of cross-compartment MSR pathways
The coordination of several MSR pathways is critical for resolving cellular stress. This is exemplified by the fact that mitochondrial membrane dynamics and mitophagy occur in conditions of UPRmt activation. Both increased fission21,118,119 and fusion27 have been observed when UPRmt is activated. Whether fission or fusion occurs presumably depends on the type and strength of the inflicting stress, thus all contributing to the proteostatic capacity and protection against spreading of damaged macromolecules and organelles. Increased mitophagy has been detected in mammalian cells and flies overexpressing mutant forms of EndoG120 or OTC-Δ (refs. 118,119,121), and upon RNAi depletion of ND75 (ref. 34), which encodes an ETC component, all conditions that strongly induce the UPRmt. Moreover, several autophagy genes are downstream targets of the master UPRmt regulator, ATFS-1, in C. elegans122. Cellular homeostasis under mitochondrial stress is hence maintained by the integrated coordination of MSR pathways. The UPRmt and presumably other interconnected protein stress responses act as the first line of defense against proteotoxicity. When the stress level overcomes the capacity of mitochondrial proteostasis, mitochondrial membrane dynamics and mitophagy come into action to redistribute and remove irreversibly damaged elements of the mitochondrial network. Inability to induce or coordinate these MSR pathways contributes to OXPHOS dysfunction and the decline in whole-organism physiology.
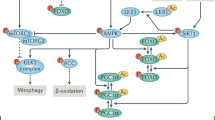
It is now evident that perturbations in the mitochondria can lead to broad cellular adaptations integrating cytosolic proteostatic responses, as shown by the recently identified mitochondrial to cytosolic stress pathways in yeast and worms123,124,125,126,127,128. These pathways restore cytosolic proteostasis by decreasing protein synthesis and/or increasing protein folding and degradation, highlighting the existence of an integrated, rather than a compartment-specific, proteostatic adaptation to mitochondrial stress. This view is supported by the integrated stress response (ISR) in mammals9. Several mitochondrial insults trigger the ISR, which induces a global response to restore cellular homeostasis by attenuating cytosolic translation and expression of cytoprotective genes. The ISR interconnects several proteostasis pathways, such as the UPR in the endoplasmic reticulum and the UPRmt, by shared signaling effectors, including eIF2α and ATF4 (refs. 9,129). Consistent with a functional link of mitochondrial to cytosolic proteostasis, pharmacological or genetic ablation of mitochondrial ribosomes attenuates cytosolic translation in worms and mammalian models9,25, induces UPRmt,, and promotes health- and lifespan extension in several organisms21,22,23 (Fig. 3a). Lipid signaling might have a coordinating role in these cross-modal stress pathways. Silencing of carnitine palmitoyltransferase (CPT) or mitochondrial chaperone mtHSP70 (hsp-6 in C. elegans) in worms triggers both the UPRmt and the heat shock response (HSR), resulting in a chaperone-mediated reduction in cytosolic proteotoxicity123. The underlying mechanism of this adaptation involves attenuation of ceramide biosynthesis123 (Fig. 3b). In line with these observations, pharmacological ceramide depletion by myriocin, a high-affinity inhibitor of the ceramide de novo biosynthesis pathway, promotes lifespan extension in yeast130 and C. elegans131. This seems to involve global proteostasis remodeling through translation attenuation and improved mitochondrial homeostasis132, common signatures of lifespan extension. By studying how yeast cells eliminate protein aggregates in the cytosol, another cross-compartment proteostasis pathway was recently revealed, wherein mitochondria can import and degrade misfolded cytosolic proteins, a phenomenon termed mitochondria as a guardian in cytosol (MAGIC)133. Further studies are needed to determine to what extent these pathways are conserved in vertebrates. One recent study, suggesting that this is indeed the case, has found that fine-tuning of mitochondrial and cytosolic translation is required for sustained killing of virally infected cells by cytotoxic T cells134.

a, Inhibition of mitochondrial translation by mrps-5 RNAi or pharmacologically using doxycycline not only blunts mitochondrial translation, but also inhibits cytosolic translation and promotes lifespan extension in C. elegans. b, In C. elegans, inhibition of CPT or silencing the mitochondrial chaperone mtHSP70 (hsp-6 in C. elegans) triggers both the UPRmt and the cytosolic HSR through a mechanism involving the attenuation of ceramide biosynthesis, termed mitochondrial to cytosolic stress response (MCSR). c, Stress signals induce mitohormesis through activation of the MSR to improve mitochondrial and cellular function. Moderate stress activates the UPRmt to prevent proteotoxicity; if the stress exceeds the capacity of the UPRmt, membrane dynamics and mitophagy are activated; sustained exposure to stress that exceeds the overall MSR capacity leads to cellular damage and organismal decline. When the amount of stress does not exceed the capacity of the MSR, the adaptive biological response leads to an improvement in cellular health and organismal lifespan.
From all this, it becomes clear that mitochondria use MSR pathways to adapt themselves and the cellular milieu to stressful situations. Thus, impairment in these MSR pathways contributes to mitochondrial dysfunction and organismal aging. Conversely, depending on the type and intensity of the stress, the MSR can lead to beneficial cellular adaptations. This finely controlled mechanism, termed mitohormesis, is proposed to protect organisms from a decline in mitochondrial function that commonly occurs during aging and to extend the lifespan across species (Fig. 3c).
mtDNA integrity in aging
mtDNA encodes only 13 OXPHOS proteins in mammals, yet it is essential for mitochondrial homeostasis. Unlike the nuclear genome, the mtDNA is replicated continuously and independently of the cell cycle. Given the inefficient mtDNA repair system, it will inevitably lead to the introduction of base errors over time. Thus, mtDNA of somatic cells is prone to accumulate mutations throughout the lifetime of an organism, which progressively leads to increasing levels of heteroplasmy, that is, the coexistence of intact and mutant mtDNA copies in the same cell. Above a certain threshold, heteroplasmy of mtDNA mutations translates into detrimental physiological consequences driving aging and disease135,136, including impairments to glucose metabolism and cognition137 and lifespan shortening138, in mice.
When mtDNA mutations occur in germline cells, they can be maternally transmitted as polymorphisms to the next generation, conferring sequence variability within species and allowing their subgroup classification as haplotypes. During evolution, mtDNA lineages, or haplogroups, emerged from the segregation of these different mtDNA sequences due to migration flow. Although enrichment of certain haplotypes might have helped our ancestors to adapt their physiology to different environmental conditions139, meta-analysis studies have reported the association of some haplotypes with several pathological conditions, such as Alzheimer’s disease140, multiple sclerosis141, and type 2 diabetes142. Conversely, other haplotypes have been associated with physiological benefits, including increased longevity in Haplogroup D among the Japanese population143 and in Haplogroup J among the European population144, although other factors, such as environment and ethnic background, could also explain this phenotype145.
Cytoplasmic hybrid (cybrid) cell lines and conplastic organismal models carrying different mtDNA variants under the same nuclear background have been extensively used to study the cellular and physiological impact of mtDNA–nDNA (in)compatibility, overcoming some limitations in association studies. In flies, mitonuclear matching has been shown to modulate lifespan146, and these epistatic interactions are further modified by diet147. Moreover, mitonuclear mismatch can have detrimental effects on mitochondrial metabolism and ROS metabolism during Drosophila aging148 and can affect their fitness149. Interestingly, a study using cybrid mouse cell lines engineered to contain different mtDNA haplotypes in an identical nuclear background showed that strain-specific mtDNA variants, such as NZB mtDNA, lead to increased ROS levels, which act as a signal for mitochondrial biogenesis150. It was later observed that conplastic mice with NZB mtDNA on the C57BL/6 nuclear background present tissue-specific reorganization of mitochondrial supercomplexes, improved ETC capacity, and improved overall energy metabolism, resulting in healthspan and lifespan extension151 (Fig. 4a). Like in the cybrid cells, this phenotype seems to involve a mitohormetic response, as these conplastic mice show a mild increase in ROS levels151, which presumably primed these beneficial adaptations. As described, a mismatch between mtDNA and nDNA can have profound consequences in physiology, which may also underpin the multivariable pathological characteristics of germline-transmitted mtDNA mutations. Owing to this haploid nature of mtDNA, high mutation rates in the mtDNA are counteracted by conserved selective mechanisms, such as bottleneck effects and purifying selection, to attenuate the transmission of deleterious levels of mutated mtDNA152,153,154,155. It has recently been shown that cells with defective mitochondria can be selectively eliminated by cell competition during early development in mice as another step of purifying selection156. Despite these protective mechanisms, heteroplasmy is often inherited, is shaped by selective forces under nuclear control in the mammalian germline157, and can affect aging. Supporting this notion, another conplastic mouse strain harboring AKR/J mtDNA under a C57BL/6 nuclear background demonstrates impairment in multiple metabolic pathways, resulting in a shorter lifespan than that of wild-type C57BL/6 mice with low levels of heteroplasmy138. Furthermore, mtDNA mutations in the maternal germline can be transmitted to mice with a wild-type nDNA background and shorten their lifespan158,159. These findings indicate that the rate of aging may be set early in life, with germline-transmitted mtDNA mutations potentially having profound lifelong consequences. With the advance of mitochondrial gene-editing techniques160, the precise manipulation of mtDNA variants may uncover how mutations in the germline affect aging and longevity.

a, The rate of aging is influenced by mitonuclear DNA matching. Mitochondrial mismatch in conplastic mice carrying NZB mtDNA under a C57BL/6 nuclear background induces ROS production, which acts as a mitohormetic signal to improve mitochondrial homeostasis, leading to lifespan extension. b, mtDNA mutations accumulate during aging by replication errors and clonal expansion of both inherited and somatic mutations. Mitochondrial fusion and mitophagy are crucial pathways involved in mtDNA integrity by diluting and eliminating mutated DNA. c, mtDNA acts as a potent DAMP, activating intracellular and extracellular pathways. In the cytosol, mtDNA stimulates the NLRP3 inflammasome, promoting IL-1β and IL-18 secretion and apoptosis by caspase-1 activation. Additionally, mtDNA is sensed by cGAS, which activates STING. This pathway triggers the IRF3 transcription factor, leading to the expression of type I IFN genes. Circulating mtDNA can elicit an immune response by activating TLR9 in endosomes, leading to NF-κB-mediated expression of IL-6 and IL-8.
The frequency of mtDNA mutations, be they point mutations or large-scale deletions, increases with age in humans and animal models161,162,163,164,165 (Fig. 4b). Deletions of mtDNA are characterized by the loss of single or multiple portions of the mitochondrial genome, which can cause both multisystemic and tissue-specific diseases42,166 Currently, it is not firmly defined whether these mutations are causal or correlative with aging, but strong indications suggest that they can contribute to OXPHOS dysfunction and some aging phenotypes167,168. This hypothesis originated from analysis of the different ‘mutator’ mouse strains39,40. The homozygous mice accumulate both point mutations and deletions in the mtDNA associated with reduced lifespan and accelerated aging, as manifested by sarcopenia, cardiomyopathy, loss of bone mass, and thymic involution39,40,169,170. Notably, over 300 human diseases are associated with POLG mutations, many of which manifest symptoms of age-related diseases171.
Conversely, although homozygous Polgmut/mut mice age prematurely, heterozygous Polgmut/+ mice seem to age normally regardless of their 500-fold higher mtDNA mutation load, suggesting that mtDNA mutation load does not define lifespan, at least up to a certain level172. This might also be true for large mtDNA deletions, as twinkle transgenic mice with multiple large mtDNA deletions do not show a reduction in the lifespan or a premature-aging phenotype173. These studies hence suggest that heteroplasmy must reach a certain level, known as the biochemical threshold174, to affect OXPHOS and organismal aging. Yet, in human tissues such as skeletal muscle, mtDNA mutations rarely reach this threshold, probably because these deletions form and clonally expand within individual muscle fibers, underlying heterogeneity across the tissue. Focal regions adjacent to myonuclei seem to be hotspots where the heteroplasmy originates before spreading across the muscle fiber175, providing evidence for a cell-specific local proliferative advantage of mutant mtDNA. These cell-specific defects could then trigger cell-nonautonomous signaling events, contributing indirectly to organismal aging.
The underlying mechanisms that govern heteroplasmy in somatic cells are largely unknown. However, it has been proposed that mutant genomes have a selective advantage over non-mutant ones, resulting in their accumulation176. For unknown reasons, the induction of the UPRmt in C. elegans seems to accelerate the accumulation of mutated mtDNA copies over intact ones48,177. In heteroplasmic worms, the UPRmt regulator ATFS-1 binds preferentially to mutated mtDNA and promotes the binding of POLG through a mechanism involving the mitochondrial protease LONP-1, suggesting that ATFS-1 stability is a key process in the maintenance of heteroplasmy178. Conversely, mitochondrial fusion and mitophagy are essential for maintaining mtDNA integrity by diluting and eliminating mutated mtDNA, respectively79,80,179,180 (Fig. 4b). Finally, mtDNA release is also a potent DAMP, activating both intracellular and extracellular immune pathways that could affect heteroplasmy (Fig. 4c). Thus a decline in MSR fitness can affect the age-related expansion of mtDNA mutations which may also have tissue181,182 and cellular specificity182,183.
Mitochondrial metabolites and aging
Nicotinamide adenine dinucleotide
NAD+ is a cofactor involved in multiple metabolic reactions. It also serves as a substrate for many NAD+-consuming enzymes, such as the poly-ADP-ribose polymerase (PARP), the cyclic ADP-ribose synthase CD38, SARM1, and sirtuins, a family of seven protein deacylases localized in the nucleus (SIRT1, SIRT6, and SIRT7), cytosol (SIRT2), and mitochondria (SIRT3–SIRT5). Decline in NAD+ levels impairs the activity of sirtuins, which are important modulators of mitochondrial homeostasis and aging184,185,186,187.
In multiple model organisms, NAD+ levels decline upon aging27,188, presumably due to reduced expression of nicotinamide phosphoribosyl (NAMPT), the rate-limiting enzyme for NAD+ synthesis189, or by increased expression and activity of NAD+-consuming enzymes, such as PARP27 and CD38 (refs. 190,191,192). In line with this view, the age-dependent decline in NAD+, which compromises mitochondrial homeostasis, can be recovered through administration of NAD+ precursors or PARP inhibition in C. elegans and mice27,188. Moreover, restoration of NAD+ levels with NR supplementation enhances lifespan in mice and improves mitochondrial and stem cell function28. These observations corroborate the protection from OXPHOS defects in mice with genetic or pharmacological ablation of PARP activity193,194, or mice treated with NMN195 or NR196. Deletor mice containing a mutation in Twinkle, which encodes a mitochondrial replicative helicase, display reduced levels of NAD+, mitochondrial impairment, and progressive muscle myopathy; treatment with NR slows early and late-stage disease progression by restoring mitochondrial function197. In agreement, both NR administration and PARP inhibition improve ETC function and exercise intolerance in Sco2-knock out/knock in mice, another model of mitochondrial disease198. Moreover, in mouse models of ataxia telangiectasia, an autosomal disorder characterized by progressive neurodegeneration and cerebellar ataxia, increasing NAD+ levels delays the accelerated-aging phenotype, including MSR decline, and extends the lifespan103. Similarly, PARP inhibition or NAD+ supplementation rescues mitochondrial dysfunction and premature aging in a mouse model of Cockayne syndrome199 and restores mitochondrial abnormalities in xeroderma pigmentosum group A (XPA)-deficient cells and worms, which prevents the attenuation in lifespan200. NAD+ depletion is also observed in people with Werner syndrome and invertebrate models of the disease, a human premature aging disease caused by mutations in the Werner DNA helicase gene102. Restoring NAD+ in C. elegans and Drosophila Werner syndrome models delays the accelerated-aging phenotype, including stem cell dysfunction, and extends the lifespan102. These findings suggest that boosting NAD+ levels prevents mitochondrial dysfunction in not only aging, but also rare genetically determined mitochondrial diseases and DNA-repair disorders known to accelerate the aging process.
Additional findings have demonstrated the importance of NAD+ in the immune system. Increasing NAD+ levels can benefit several inflammatory conditions in mouse models of aging28,188, ataxia-telangiectasia autoimmunity103, and muscular dystrophy51. In older humans, NR administration for only 21 days was sufficient to reduce circulating inflammatory cytokines201. However, it is unclear whether this anti-inflammatory effect is secondary to the physiological benefits of NAD+ or perhaps is more probably caused by direct programming of immune cells. NAD+ levels decline in immune cells upon aging, and boosting NAD+ levels restores the age-related decrease in OXPHOS and immune function in macrophages from older humans and mice202. Interestingly, pro-inflammatory M1-like, but not naive or M2, macrophages express high levels of the NAD-consuming enzyme CD38, induced by cytokines released from senescent cells190,191. These M1-like macrophages accumulate in tissues such as visceral white adipose tissue and liver during aging, thereby reducing global tissue NAD+ levels191, suggesting that senescent cells promote tissue NAD+ reduction via activation of macrophages.
Collectively, these findings suggest that a decrease in systemic NAD+ levels is a crucial driver of organismal decline in aging. This is further supported by the overarching therapeutic effect of NAD+ boosters in several animal models of common age-related conditions, ranging from diabetes and obesity195,196, non-alcoholic fatty liver disease203, kidney injury204,205,206, impaired muscle function and sarcopenia51,188,196, glaucoma207, ischemia–reperfusion injury208,209, vascular dysfunction210, to cognitive decline56,103,211,212. Taken together, the health benefits and the prevention of age-associated MSR and OXPHOS decline support the use of NAD+ boosters as therapy for some of these age-related diseases. The efficacy of NAD replenishment was recently illustrated in the setting of human acute kidney injury206 and COVID-19 (ref. 213).
Tricarboxylic acid cycle intermediates
Tricarboxylic acid cycle (TCA) metabolites are by-products of energy metabolism with essential roles in cellular homeostasis, fueling anabolic reactions and adjusting metabolic pathways through signaling cascades or allosteric modulation of key enzymes. There is increasing evidence that TCA metabolites are essential mediators of cellular signaling by their actions in chromatin modifications, DNA methylation, and post-translational protein modifications214. For instance, citrate leads to the cytosolic production of acetyl-CoA that fuels histone and protein acetylation through acetyltransferases215,216, thus modulating gene expression217,218,219,220. In this regard, impaired mitochondrial metabolism affects epigenetics by restricting the production of TCA intermediates. Indeed, genetic ablation of the ETC impairs histone acetylation, which can be restored by reconstitution of TCA function in human cells221. Moreover, DNA- and histone-methylation status are both regulated by 2-oxoglutarate-dependent dioxygenases (2-OGDO), such as ten-eleven translocation (TET) hydroxylases and histone demethylases that use α-ketoglutarate (α-KG) and oxygen as co-substrates for oxidation of target molecules214. Succinate and fumarate are potent inhibitors of these 2-OGDO enzymes222, underscoring the tight control of the chromatin epigenetic landscape by the TCA.
Changes in nutritional pathways and epigenetic state are crucial to aging and are affected by TCA metabolites. In C. elegans and Drosophila, administration of α-KG extends lifespan through a mechanism involving the inhibition of the target of rapamycin (TOR)223,224. Fumarate and malate, when administered to worms, extend lifespan, which is associated with the induction of the glyoxylate shunt, an extra-mitochondrial pathway of energy production, mild mitochondrial uncoupling, and expression of the longevity regulators DAF-16 and SIR-2.1 (refs. 225,226). Conversely, the accumulation of succinate causes an opposite effect in worms and flies: succinate dehydrogenase (SDH) mutants display increased ROS levels and accelerated aging227,228. Acetyl-CoA seems to be an essential mitochondrial signal regulating the rate of aging in C. elegans. Upon mitochondrial stress early in life, levels of acetyl-CoA decrease, resulting in the nuclear accumulation of the histone deacetylase complex (NuRD), allowing epigenetic and transcriptional remodeling for lifespan extension in worms229.
α-KG levels decline upon mammalian aging230,231 and correlate with alterations in the epigenetic landscape in several tissues, such as the brain232, adipose tissue231, and bones233. Not surprisingly, replenishment of α-KG levels attenuates several age-related disorders. Increasing the levels of α-KG ameliorates age-related osteoporosis in aged mice by reducing accumulation of histone H3 trimethylated at K9 and H3 trimethylated at K27 at the promoters of the osteogenesis-related genes Bmp2, Bmp4, and Nanog, improving bone marrow mesenchymal stromal and stem cell function233. Moreover, restoration of α-KG in middle-aged mice increases DNA demethylation at the promoter of the transcriptional regulator of brown adipocytes Prdm16, which induces brown adipocyte genes and prevents age-associated obesity231. Increasing α-KG levels furthermore restores age-related redox alterations234, reduces inflammation235, delays fertility decline236, and extends the lifespan235 in mice (as in flies and worms—see above), suggesting that α-KG is a potent signaling metabolic intermediate involved in mammalian aging, presumably by epigenetic modulation. Also, an elevated α-KG:succinate ratio is involved in the maintenance and differentiation of pluripotency of embryonic stem cells237,238 and modulates the differentiation of germ cells239 by DNA and histone demethylation. Similar to what happens in C. elegans, accumulation of succinate by reduced SDH activity in mammals, as observed during aging240,241, can counteract the epigenetic actions of α-KG by inhibiting 2-OGDO demethylases237,238, thus contributing to age-related epigenetic alterations232 and diseases. Fumarate has recently been shown to act as a terminal electron acceptor in the mammalian ETC under conditions of hypoxia, yet its connection with aging remains to be established in mammals242.
Conclusion and perspectives
Work over recent years has uncovered the impressive ability of the mitochondria to maintain homeostasis in a variety of stressful situations. The importance of this adaptive response is underlined by our increasing understanding of how defects in these mitochondrial responses are intimately associated with aging. Mitochondria have a pleiotropic effect on aging, which can comprise protective or maladaptive responses. The nature of their response will depend on how mitochondria can sustain their MSR pathways within the ever-changing cellular milieu that is exposing them constantly to various levels of stress (Fig. 5). In recent years, we have gained considerable insight into how age-related processes are intimately wired to different types of MSR. Still, the functional interactions of these pathways and their implication in aging remain largely unexplored (Box 2). Identifying shared molecular signals of stress responses will be crucial to shed light on how MSR dysfunction contributes to proteostasis collapse during the aging process243. For instance, NAD+ gradually declines during aging and seems to integrate many of these stress responses. Other signaling molecules, such as bioactive lipids and mitochondrial metabolites, may warrant more attention, as they also seem to have a role in cross-compartment stress communication. On the same note, mitochondrial DNA has evolved as a critical signaling factor in cellular homeostasis. Given some unique features of mitochondria, such as their haploid inheritance, constant replication rates, and inefficient DNA-repair system, mtDNA is prone to accumulating mutations throughout life, leading to a progressive increase in heteroplasmy. Moreover, the rate of aging may be set early in life by germline-transmitted mtDNA mutations. Yet, the underlying mechanisms in mammals are poorly known. Future studies should focus on the involvement of MSR in regulating the clonal expansion of both somatic and inherited mtDNA mutations, as observed in model organisms69,82. Under these circumstances, how damaged mitochondria or components of the mitochondria, including mtDNA, are released during aging and whether this signals cellular and systemic inflammation are important questions that should be addressed. Hopefully, unveiling the pleiotropic effects of mitochondrial dysfunction will allow us to better understand fundamental aspects of how mitochondria have a commanding role in the aging clock.

Mitochondrial alterations in aging initiate with a decline in MSR pathways leading to the accumulation of mtDNA mutations, release of damaged toxic mitochondrial material (for example, DAMPs), mtROS generation, proteotoxicity, and deregulated metabolites (TCA intermediates, NAD+). These alterations have a broad detrimental effect on cellular homeostasis and, through a complex signaling mechanism (involving mitokines, metabolites, and more), contribute to systemic organismal decline and the onset of several age-related diseases. Pharmacological modulation of the MSR, such as through the use of NAD+ enhancers or mitophagy inducers, can be effective strategies to prevent aging-related cellular and organismal decline. IR injury, ischemia–reperfusion injury; NFLD, non-alcoholic fatty liver disease; NASH, non-alcoholic steatohepatitis.
References
Sagan, L. On the origin of mitosing cells. J. Theor. Biol. 14, 255–274 (1967).
Ryan, M. T. & Hoogenraad, N. J. Mitochondrial–nuclear communications. Annu. Rev. Biochem. 76, 701–722 (2007).
Quirós, P. M., Mottis, A. & Auwerx, J. Mitonuclear communication in homeostasis and stress. Nat. Rev. Mol. Cell Biol. 17, 213–226 (2016).
Ng, M. Y. W., Wai, T. & Simonsen, A. Quality control of the mitochondrion. Dev. Cell 56, 881–905 (2021).
Sun, N., Youle, R. J. & Finkel, T. The mitochondrial basis of aging. Mol. Cell 61, 654–666 (2016).
Schieber, M. & Chandel, N. S. ROS function in redox signaling and oxidative stress. Curr. Biol. 24, R453–62 (2014).
Zhang, H., Menzies, K. J. & Auwerx, J. The role of mitochondria in stem cell fate and aging. Development 145, dev143420 (2018).
Tatsuta, T. & Langer, T. Quality control of mitochondria: protection against neurodegeneration and ageing. EMBO J. 27, 306–314 (2008).
Quirós, P. M. et al. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol. 216, 2027–2045 (2017).
Zhao, Q. et al. A mitochondrial specific stress response in mammalian cells. EMBO J. 21, 4411–4419 (2002).
Fiorese, C. J. et al. The transcription factor ATF5 mediates a mammalian mitochondrial UPR. Curr. Biol. 26, 2037–2043 (2016).
Nargund, A. M., Pellegrino, M. W., Fiorese, C. J., Baker, B. M. & Haynes, C. M. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 337, 587–590 (2012).
Copeland, J. M. et al. Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Curr. Biol. 19, 1591–1598 (2009).
Dell’agnello, C. et al. Increased longevity and refractoriness to Ca2+-dependent neurodegeneration in Surf1 knockout mice. Hum. Mol. Genet. 16, 431–444 (2007).
Feng, J., Bussière, F. & Hekimi, S. Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans. Dev. Cell 1, 633–644 (2001).
Liu, X. et al. Evolutionary conservation of the clk-1-dependent mechanism of longevity: loss of mclk1 increases cellular fitness and lifespan in mice. Genes Dev. 19, 2424–2434 (2005).
Lee, S. S. et al. A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat. Genet. 33, 40–48 (2003).
Dillin, A. et al. Rates of behavior and aging specified by mitochondrial function during development. Science 298, 2398–2401 (2002).
Kuang, J. & Ebert, P. R. The failure to extend lifespan via disruption of complex II is linked to preservation of dynamic control of energy metabolism. Mitochondrion 12, 280–287 (2012).
Pulliam, D. A. et al. Complex IV-deficient Surf1–/– mice initiate mitochondrial stress responses. Biochem. J. 462, 359–371 (2014).
Houtkooper, R. H. et al. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 497, 451–457 (2013).
Wang, X. & Auwerx, J. Systems phytohormone responses to mitochondrial proteotoxic stress. Mol. Cell 68, 540–551 (2017).
Moullan, N. et al. Tetracyclines disturb mitochondrial function across eukaryotic models: a call for caution in biomedical research. Cell Rep. 10, 1681–1691 (2015).
Suhm, T. et al. Mitochondrial translation efficiency controls cytoplasmic protein homeostasis. Cell Metab. 27, 1309–1322 (2018).
Molenaars, M. et al. A conserved mito-cytosolic translational balance links two longevity pathways. Cell Metab. 31, 549–563 (2020).
D’Amico, D., Sorrentino, V. & Auwerx, J. Cytosolic proteostasis networks of the mitochondrial stress response. Trends Biochem. Sci. 42, 712–725 (2017).
Mouchiroud, L. et al. The NAD+/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell 154, 430–441 (2013).
Zhang, H. et al. NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 352, 1436–1443 (2016).
Harrison, D. E. et al. 17-α-estradiol late in life extends lifespan in aging UM-HET3 male mice; nicotinamide riboside and three other drugs do not affect lifespan in either sex. Aging Cell 20, e13328 (2021).
Durieux, J., Wolff, S. & Dillin, A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 144, 79–91 (2011).
Berendzen, K. M. et al. Neuroendocrine coordination of mitochondrial stress signaling and proteostasis. Cell 166, 1553–1563 (2016).
Zhang, Q. et al. The mitochondrial unfolded protein response is mediated cell-non-autonomously by retromer-dependent Wnt signaling. Cell 174, 870–883 (2018).
Zhang, Q. et al. The memory of neuronal mitochondrial stress is inherited transgenerationally via elevated mitochondrial DNA levels. Nat. Cell Biol. 23, 870–880 (2021).
Owusu-Ansah, E., Song, W. & Perrimon, N. Muscle mitohormesis promotes longevity via systemic repression of insulin signaling. Cell 155, 699–712 (2013).
Kang, S. G. et al. Differential roles of GDF15 and FGF21 in systemic metabolic adaptation to the mitochondrial integrated stress response. iScience 24, 102181 (2021).
Chung, H. K. et al. Growth differentiation factor 15 is a myomitokine governing systemic energy homeostasis. J. Cell Biol. 216, 149–165 (2017).
Wang, X. et al. hNAG-1 increases lifespan by regulating energy metabolism and insulin/IGF-1/mTOR signaling. Aging 6, 690–704 (2014).
Zhang, Y. et al. The starvation hormone, fibroblast growth factor-21, extends lifespan in mice. eLife 1, e00065 (2012).
Kujoth, G. C. et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 309, 481–484 (2005).
Trifunovic, A. et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429, 417–423 (2004).
Kruse, S. E. et al. Mice with mitochondrial complex I deficiency develop a fatal encephalomyopathy. Cell Metab. 7, 312–320 (2008).
Gorman, G. S. et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 77, 753–759 (2015).
Lake, N. J., Bird, M. J., Isohanni, P. & Paetau, A. Leigh syndrome: neuropathology and pathogenesis. J. Neuropathol. Exp. Neurol. 74, 482–492 (2015).
Forsström, S. et al. Fibroblast growth factor 21 drives dynamics of local and systemic stress responses in mitochondrial myopathy with mtDNA deletions. Cell Metab. 30, 1040–1054 (2019).
Lehtonen, J. M. et al. FGF21 is a biomarker for mitochondrial translation and mtDNA maintenance disorders. Neurology 87, 2290–2299 (2016).
Suomalainen, A. et al. FGF-21 as a biomarker for muscle-manifesting mitochondrial respiratory chain deficiencies: a diagnostic study. Lancet Neurol. 10, 806–818 (2011).
Conte, M. et al. Human aging and longevity are characterized by high levels of mitokines. J. Gerontol. A Biol. Sci. Med. Sci. 74, 600–607 (2019).
Lin, Y.-F. et al. Maintenance and propagation of a deleterious mitochondrial genome by the mitochondrial unfolded protein response. Nature 533, 416–419 (2016).
Khan, N. A. et al. mTORC1 regulates mitochondrial integrated stress response and mitochondrial myopathy progression. Cell Metab. 26, 419–428.e5 (2017).
Ma, C. et al. Nampt expression decreases age-related senescence in rat bone marrow mesenchymal stem cells by targeting Sirt1. PLoS ONE 12, e0170930 (2017).
Ryu, D. et al. NAD+ repletion improves muscle function in muscular dystrophy and counters global PARylation. Sci. Transl. Med. 8, 361ra139 (2016).
Ryu, D. et al. A SIRT7-dependent acetylation switch of GABPβ1 controls mitochondrial function. Cell Metab. 20, 856–869 (2014).
Mohrin, M. et al. Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science 347, 1374–1377 (2015).
Wu, Y. et al. Multilayered genetic and omics dissection of mitochondrial activity in a mouse reference population. Cell 158, 1415–1430 (2014).
Migliavacca, E. et al. Mitochondrial oxidative capacity and NAD+ biosynthesis are reduced in human sarcopenia across ethnicities. Nat. Commun. 10, 5808 (2019).
Sorrentino, V. et al. Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity. Nature 552, 187–193 (2017).
Beck, J. S., Mufson, E. J. & Counts, S. E. Evidence for mitochondrial UPR gene activation in familial and sporadic Alzheimer’s disease. Curr. Alzheimer Res. 13, 610–614 (2016).
Collins, T. J., Berridge, M. J., Lipp, P. & Bootman, M. D. Mitochondria are morphologically and functionally heterogeneous within cells. EMBO J. 21, 1616–1627 (2002).
Eisner, V., Picard, M. & Hajnóczky, G. Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat. Cell Biol. 20, 755–765 (2018).
Youle, R. J. & van der Bliek, A. M. Mitochondrial fission, fusion, and stress. Science 337, 1062–1065 (2012).
Giacomello, M., Pyakurel, A., Glytsou, C. & Scorrano, L. The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol. 21, 204–224 (2020).
Kraus, F., Roy, K., Pucadyil, T. J. & Ryan, M. T. Function and regulation of the divisome for mitochondrial fission. Nature 590, 57–66 (2021).
Twig, G. et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 27, 433–446 (2008).
Kleele, T. et al. Distinct fission signatures predict mitochondrial degradation or biogenesis. Nature 593, 435–439 (2021).
Scheckhuber, C. Q. et al. Reducing mitochondrial fission results in increased life span and fitness of two fungal ageing models. Nat. Cell Biol. 9, 99–105 (2007).
Jiang, H.-C. et al. Neural activity and CaMKII protect mitochondria from fragmentation in aging Caenorhabditis elegans neurons. Proc. Natl Acad. Sci. USA 112, 8768–8773 (2015).
D’Amico, D. et al. The RNA-binding protein PUM2 impairs mitochondrial dynamics and mitophagy during aging. Mol. Cell 73, 775–787 (2019).
Chaudhari, S. N. & Kipreos, E. T. Increased mitochondrial fusion allows the survival of older animals in diverse C. elegans longevity pathways. Nat. Commun. 8, 182 (2017).
Bohnert, K. A. & Kenyon, C. A lysosomal switch triggers proteostasis renewal in the immortal C. elegans germ lineage. Nature 551, 629–633 (2017).
Rana, A., Rera, M. & Walker, D. W. Parkin overexpression during aging reduces proteotoxicity, alters mitochondrial dynamics, and extends lifespan. Proc. Natl Acad. Sci. USA 110, 8638–8643 (2013).
Rana, A. et al. Promoting Drp1-mediated mitochondrial fission in midlife prolongs healthy lifespan of Drosophila melanogaster. Nat. Commun. 8, 448 (2017).
Liu, Y. J. et al. Mitochondrial translation and dynamics synergistically extend lifespan in C. elegans through HLH-30. J. Cell Biol. 219, e201907067 (2020).
Bernhardt, D., Müller, M., Reichert, A. S. & Osiewacz, H. D. Simultaneous impairment of mitochondrial fission and fusion reduces mitophagy and shortens replicative lifespan. Sci. Rep. 5, 7885 (2015).
Weir, H. J. et al. Dietary restriction and AMPK increase lifespan via mitochondrial network and peroxisome remodeling. Cell Metab. 26, 884–896.e5 (2017).
Udagawa, O. et al. Mitochondrial fission factor Drp1 maintains oocyte quality via dynamic rearrangement of multiple organelles. Curr. Biol. 24, 2451–2458 (2014).
Kageyama, Y. et al. Mitochondrial division ensures the survival of postmitotic neurons by suppressing oxidative damage. J. Cell Biol. 197, 535–551 (2012).
Dulac, M. et al. Regulation of muscle and mitochondrial health by the mitochondrial fission protein Drp1 in aged mice. J. Physiol. 599, 4045–4063 (2021).
Song, M., Franco, A., Fleischer, J. A., Zhang, L. & Dorn, G. W. Abrogating mitochondrial dynamics in mouse hearts accelerates mitochondrial senescence. Cell Metab. 26, 872–883 (2017).
Chen, H. et al. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 141, 280–289 (2010).
Amati-Bonneau, P. et al. OPA1 mutations induce mitochondrial DNA instability and optic atrophy “plus” phenotypes. Brain 131, 338–351 (2008).
Hudson, G. et al. Mutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: a novel disorder of mtDNA maintenance. Brain 131, 329–337 (2008).
Lieber, T., Jeedigunta, S. P., Palozzi, J. M., Lehmann, R. & Hurd, T. R. Mitochondrial fragmentation drives selective removal of deleterious mtDNA in the germline. Nature 570, 380–384 (2019).
Pickles, S., Vigié, P. & Youle, R. J. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. 28, R170–R185 (2018).
Hansen, M., Rubinsztein, D. C. & Walker, D. W. Autophagy as a promoter of longevity: insights from model organisms. Nat. Rev. Mol. Cell Biol. 19, 579–593 (2018).
Palikaras, K., Lionaki, E. & Tavernarakis, N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 521, 525–528 (2015).
Schiavi, A. et al. Iron-starvation-induced mitophagy mediates lifespan extension upon mitochondrial stress in C. elegans. Curr. Biol. 25, 1810–1822 (2015).
Greene, J. C. et al. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc. Natl Acad. Sci. USA 100, 4078–4083 (2003).
Sun, N. et al. Measuring in vivo mitophagy. Mol. Cell 60, 685–696 (2015).
Pickrell, A. M. et al. Endogenous parkin preserves dopaminergic substantia nigral neurons following mitochondrial DNA mutagenic stress. Neuron 87, 371–381 (2015).
Hoshino, A. et al. Cytosolic p53 inhibits parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat. Commun. 4, 2308 (2013).
García-Prat, L. et al. Autophagy maintains stemness by preventing senescence. Nature 529, 37–42 (2016).
Ryu, D. et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 22, 879–888 (2016).
Luan, P. et al. Urolithin A improves muscle function by inducing mitophagy in muscular dystrophy. Sci. Transl. Med. 13, eabb0319 (2021).
Zhang, H. et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 283, 10892–10903 (2008).
Jain, I. H. et al. Hypoxia as a therapy for mitochondrial disease. Science 352, 54–61 (2016).
Zhang, W. et al. Hypoxic mitophagy regulates mitochondrial quality and platelet activation and determines severity of I/R heart injury. eLife 5, e21407 (2016).
Andreux, P. A. et al. The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nat. Metab. 1, 595–603 (2019).
Fang, E. F. et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 22, 401–412 (2019).
D’Amico, D. et al. Impact of the natural compound urolithin A on health, disease, and aging. Trends Mol. Med. 27, 687–699 (2021).
Schwarz, C. et al. Safety and tolerability of spermidine supplementation in mice and older adults with subjective cognitive decline. Aging 10, 19–33 (2018).
Madeo, F., Eisenberg, T., Pietrocola, F. & Kroemer, G. Spermidine in health and disease. Science 359, eaan2788 (2018).
Fang, E. F. et al. NAD+ augmentation restores mitophagy and limits accelerated aging in Werner syndrome. Nat. Commun. 10, 5284 (2019).
Fang, E. F. et al. NAD+ replenishment improves lifespan and healthspan in ataxia telangiectasia models via mitophagy and DNA repair. Cell Metab. 24, 566–581 (2016).
Frank-Cannon, T. C. et al. Parkin deficiency increases vulnerability to inflammation-related nigral degeneration. J. Neurosci. 28, 10825–10834 (2008).
Sliter, D. A. et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 561, 258–262 (2018).
Matheoud, D. et al. Intestinal infection triggers Parkinson’s disease-like symptoms in Pink1−/− mice. Nature 571, 565–569 (2019).
Matheoud, D. et al. Parkinson’s disease-related proteins PINK1 and parkin repress mitochondrial antigen presentation. Cell 166, 314–327 (2016).
Franceschi, C., Garagnani, P., Parini, P., Giuliani, C. & Santoro, A. Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 14, 576–590 (2018).
Mills, E. L., Kelly, B. & O’Neill, L. A. J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 18, 488–498 (2017).
Nakahira, K. et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 12, 222–230 (2011).
Zhong, Z. et al. NF-κB restricts inflammasome activation via elimination of damaged mitochondria. Cell 164, 896–910 (2016).
Rai, P. et al. IRGM1 links mitochondrial quality control to autoimmunity. Nat. Immunol. 22, 312–321 (2021).
Patoli, D. et al. Inhibition of mitophagy drives macrophage activation and antibacterial defense during sepsis. J. Clin. Invest. 130, 5858–5874 (2020).
Pinti, M. et al. Circulating mitochondrial DNA increases with age and is a familiar trait: implications for “inflamm-aging”. Eur. J. Immunol. 44, 1552–1562 (2014).
Rodríguez-Nuevo, A. et al. Mitochondrial DNA and TLR9 drive muscle inflammation upon Opa1 deficiency. EMBO J. 37, e96553 (2018).
Al Amir Dache, Z. et al. Blood contains circulating cell-free respiratory competent mitochondria. FASEB J. 34, 3616–3630 (2020).
Trumpff, C. et al. Stress and circulating cell-free mitochondrial DNA: a systematic review of human studies, physiological considerations, and technical recommendations. Mitochondrion 59, 225–245 (2021).
Burbulla, L. F. et al. Mitochondrial proteolytic stress induced by loss of mortalin function is rescued by parkin and PINK1. Cell Death Dis. 5, e1180 (2014).
Pimenta de Castro, I. et al. Genetic analysis of mitochondrial protein misfolding in Drosophila melanogaster. Cell Death Differ. 19, 1308–1316 (2012).
Papa, L. & Germain, D. Sirt3 regulates the mitochondrial unfolded protein response. Mol. Cell. Biol. 34, 699–710 (2014).
Jin, S. M. & Youle, R. J. The accumulation of misfolded proteins in the mitochondrial matrix is sensed by PINK1 to induce PARK2/parkin-mediated mitophagy of polarized mitochondria. Autophagy 9, 1750–1757 (2013).
Guo, B. et al. Genome-wide screen identifies signaling pathways that regulate autophagy during Caenorhabditis elegans development. EMBO Rep. 15, 705–713 (2014).
Kim, H.-E. et al. Lipid biosynthesis coordinates a mitochondrial-to-cytosolic stress response. Cell 166, 1539–1552 (2016).
Mårtensson, C. U. et al. Mitochondrial protein translocation-associated degradation. Nature 569, 679–683 (2019).
Weidberg, H. & Amon, A. MitoCPR-A surveillance pathway that protects mitochondria in response to protein import stress. Science 360, eaan4146 (2018).
Wrobel, L. et al. Mistargeted mitochondrial proteins activate a proteostatic response in the cytosol. Nature 524, 485–488 (2015).
Wang, X. & Chen, X. J. A cytosolic network suppressing mitochondria-mediated proteostatic stress and cell death. Nature 524, 481–484 (2015).
Boos, F. et al. Mitochondrial protein-induced stress triggers a global adaptive transcriptional programme. Nat. Cell Biol. 21, 442–451 (2019).
Frakes, A. E. & Dillin, A. The UPRER: sensor and coordinator of organismal homeostasis. Mol. Cell 66, 761–771 (2017).
Huang, X., Liu, J. & Dickson, R. C. Down-regulating sphingolipid synthesis increases yeast lifespan. PLoS Genet. 8, e1002493 (2012).
Cutler, R. G., Thompson, K. W., Camandola, S., Mack, K. T. & Mattson, M. P. Sphingolipid metabolism regulates development and lifespan in Caenorhabditis elegans. Mech. Ageing Dev. 143–144, 9–18 (2014).
Liu, J. et al. Reducing sphingolipid synthesis orchestrates global changes to extend yeast lifespan. Aging Cell 12, 833–841 (2013).
Ruan, L. et al. Cytosolic proteostasis through importing of misfolded proteins into mitochondria. Nature 543, 443–446 (2017).
Lisci, M. et al. Mitochondrial translation is required for sustained killing by cytotoxic T cells. Science 374, eabe9977 (2021).
Kauppila, T. E. S., Kauppila, J. H. K. & Larsson, N.-G. Mammalian mitochondria and aging: an update. Cell Metab. 25, 57–71 (2017).
Stewart, J. B. & Chinnery, P. F. The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat. Rev. Genet. 16, 530–542 (2015).
Sharpley, M. S. et al. Heteroplasmy of mouse mtDNA is genetically unstable and results in altered behavior and cognition. Cell 151, 333–343 (2012).
Hirose, M. et al. Low-level mitochondrial heteroplasmy modulates DNA replication, glucose metabolism and lifespan in mice. Sci. Rep. 8, 5872 (2018).
Ruiz-Pesini, E., Mishmar, D., Brandon, M., Procaccio, V. & Wallace, D. C. Effects of purifying and adaptive selection on regional variation in human mtDNA. Science 303, 223–226 (2004).
Bi, R. et al. Mitochondrial DNA haplogroup B5 confers genetic susceptibility to Alzheimer’s disease in Han Chinese. Neurobiol. Aging 36, 1604.e7–16 (2015).
Tranah, G. J. et al. Mitochondrial DNA sequence variation in multiple sclerosis. Neurology 85, 325–330 (2015).
Chinnery, P. F. et al. Mitochondrial DNA haplogroups and type 2 diabetes: a study of 897 cases and 1010 controls. J. Med. Genet. 44, e80 (2007).
Tanaka, M., Gong, J. S., Zhang, J., Yoneda, M. & Yagi, K. Mitochondrial genotype associated with longevity. Lancet 351, 185–186 (1998).
De Benedictis, G. et al. Mitochondrial DNA inherited variants are associated with successful aging and longevity in humans. FASEB J. 13, 1532–1536 (1999).
Pinós, T. et al. Are mitochondrial haplogroups associated with extreme longevity? A study on a Spanish cohort. Age 34, 227–233 (2012).
Rand, D. M., Fry, A. & Sheldahl, L. Nuclear-mitochondrial epistasis and drosophila aging: introgression of Drosophila simulans mtDNA modifies longevity in D. melanogaster nuclear backgrounds. Genetics 172, 329–341 (2006).
Zhu, C.-T., Ingelmo, P. & Rand, D. M. G×G×E for lifespan in Drosophila: mitochondrial, nuclear, and dietary interactions that modify longevity. PLoS Genet. 10, e1004354 (2014).
Pichaud, N. et al. Age dependent dysfunction of mitochondrial and ROS metabolism induced by mitonuclear mismatch. Front. Genet. 10, 130 (2019).
Sujkowski, A. et al. Mito-nuclear interactions modify Drosophila exercise performance. Mitochondrion 47, 188–205 (2019).
Moreno-Loshuertos, R. et al. Differences in reactive oxygen species production explain the phenotypes associated with common mouse mitochondrial DNA variants. Nat. Genet. 38, 1261–1268 (2006).
Latorre-Pellicer, A. et al. Mitochondrial and nuclear DNA matching shapes metabolism and healthy ageing. Nature 535, 561–565 (2016).
Stewart, J. B. et al. Strong purifying selection in transmission of mammalian mitochondrial DNA. PLoS Biol. 6, e10 (2008).
Fan, W. et al. A mouse model of mitochondrial disease reveals germline selection against severe mtDNA mutations. Science 319, 958–962 (2008).
Freyer, C. et al. Variation in germline mtDNA heteroplasmy is determined prenatally but modified during subsequent transmission. Nat. Genet. 44, 1282–1285 (2012).
Floros, V. I. et al. Segregation of mitochondrial DNA heteroplasmy through a developmental genetic bottleneck in human embryos. Nat. Cell Biol. 20, 144–151 (2018).
Lima, A. et al. Cell competition acts as a purifying selection to eliminate cells with mitochondrial defects during early mouse development. Nat. Metab. 3, 1091–1108 (2021).
Wei, W. et al. Germline selection shapes human mitochondrial DNA diversity. Science 364, eaau6520 (2019).
Ross, J. M. et al. Germline mitochondrial DNA mutations aggravate ageing and can impair brain development. Nature 501, 412–415 (2013).
Ross, J. M., Coppotelli, G., Hoffer, B. J. & Olson, L. Maternally transmitted mitochondrial DNA mutations can reduce lifespan. Sci. Rep. 4, 6569 (2014).
Mok, B. Y. et al. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature 583, 631–637 (2020).
Pikó, L., Hougham, A. J. & Bulpitt, K. J. Studies of sequence heterogeneity of mitochondrial DNA from rat and mouse tissues: evidence for an increased frequency of deletions/additions with aging. Mech. Ageing Dev. 43, 279–293 (1988).
Greaves, L. C. et al. Clonal expansion of early to mid-life mitochondrial DNA point mutations drives mitochondrial dysfunction during human ageing. PLoS Genet. 10, e1004620 (2014).
Meissner, C. et al. The 4977 bp deletion of mitochondrial DNA in human skeletal muscle, heart and different areas of the brain: a useful biomarker or more? Exp. Gerontol. 43, 645–652 (2008).
Zhang, R., Wang, Y., Ye, K., Picard, M. & Gu, Z. Independent impacts of aging on mitochondrial DNA quantity and quality in humans. BMC Genomics 18, 890 (2017).
Herbst, A. et al. Mitochondrial DNA deletion mutations increase exponentially with age in human skeletal muscle. Aging Clin. Exp. Res. 33, 1811–1820 (2021).
Oldfors, A. et al. Mitochondrial DNA deletions in muscle fibers in inclusion body myositis. J. Neuropathol. Exp. Neurol. 54, 581–587 (1995).
Larsson, N.-G. Somatic mitochondrial DNA mutations in mammalian aging. Annu. Rev. Biochem. 79, 683–706 (2010).
Payne, B. A. I. & Chinnery, P. F. Mitochondrial dysfunction in aging: much progress but many unresolved questions. Biochim. Biophys. Acta 1847, 1347–1353 (2015).
Zhang, D. et al. Mitochondrial DNA mutations activate the mitochondrial apoptotic pathway and cause dilated cardiomyopathy. Cardiovasc. Res. 57, 147–157 (2003).
Trifunovic, A. et al. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc. Natl Acad. Sci. USA 102, 17993–17998 (2005).
DeBalsi, K. L., Hoff, K. E. & Copeland, W. C. Role of the mitochondrial DNA replication machinery in mitochondrial DNA mutagenesis, aging and age-related diseases. Ageing Res. Rev. 33, 89–104 (2017).
Vermulst, M. et al. Mitochondrial point mutations do not limit the natural lifespan of mice. Nat. Genet. 39, 540–543 (2007).
Tyynismaa, H. et al. Mutant mitochondrial helicase Twinkle causes multiple mtDNA deletions and a late-onset mitochondrial disease in mice. Proc. Natl Acad. Sci. USA 102, 17687–17692 (2005).
Rossignol, R. et al. Mitochondrial threshold effects. Biochem. J. 370, 751–762 (2003).
Vincent, A. E. et al. Subcellular origin of mitochondrial DNA deletions in human skeletal muscle. Ann. Neurol. 84, 289–301 (2018).
Chinnery, P. F., Samuels, D. C., Elson, J. & Turnbull, D. M. Accumulation of mitochondrial DNA mutations in ageing, cancer, and mitochondrial disease: is there a common mechanism? Lancet 360, 1323–1325 (2002).
Gitschlag, B. L. et al. Homeostatic responses regulate selfish mitochondrial genome dynamics in C. elegans. Cell Metab. 24, 91–103 (2016).
Yang, Q. et al. LONP-1 and ATFS-1 sustain deleterious heteroplasmy by promoting mtDNA replication in dysfunctional mitochondria. Nat. Cell Biol. 28, 181–193 (2022).
Kandul, N. P., Zhang, T., Hay, B. A. & Guo, M. Selective removal of deletion-bearing mitochondrial DNA in heteroplasmic Drosophila. Nat. Commun. 7, 13100 (2016).
Suen, D.-F., Narendra, D. P., Tanaka, A., Manfredi, G. & Youle, R. J. Parkin overexpression selects against a deleterious mtDNA mutation in heteroplasmic cybrid cells. Proc. Natl Acad. Sci. USA 107, 11835–11840 (2010).
Jenuth, J. P., Peterson, A. C. & Shoubridge, E. A. Tissue-specific selection for different mtDNA genotypes in heteroplasmic mice. Nat. Genet. 16, 93–95 (1997).
Hämäläinen, R. H. et al. Tissue- and cell-type-specific manifestations of heteroplasmic mtDNA 3243A>G mutation in human induced pluripotent stem cell-derived disease model. Proc. Natl Acad. Sci. USA 110, E3622–30 (2013).
Walker, M. A. et al. Purifying selection against pathogenic mitochondrial DNA in human T cells. N. Engl. J. Med. 383, 1556–1563 (2020).
Houtkooper, R. H., Pirinen, E. & Auwerx, J. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 13, 225–238 (2012).
Katsyuba, E., Romani, M., Hofer, D. & Auwerx, J. NAD+ homeostasis in health and disease. Nat. Metab. 2, 9–31 (2020).
Rajman, L., Chwalek, K. & Sinclair, D. A. Therapeutic potential of NAD-boosting molecules: the in vivo evidence. Cell Metab. 27, 529–547 (2018).
Imai, S.-I. & Guarente, L. It takes two to tango: NAD+ and sirtuins in aging/longevity control. npj Aging Mech. Dis. 2, 16017 (2016).
Gomes, A. P. et al. Declining NAD+ induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 155, 1624–1638 (2013).
Nakahata, Y., Sahar, S., Astarita, G., Kaluzova, M. & Sassone-Corsi, P. Circadian control of the NAD+ salvage pathway by CLOCK–SIRT1. Science 324, 654–657 (2009).
Covarrubias, A. J. et al. Senescent cells promote tissue NAD+ decline during ageing via the activation of CD38+ macrophages. Nat. Metab. 2, 1265–1283 (2020).
Chini, C. C. S. et al. CD38 ecto-enzyme in immune cells is induced during aging and regulates NAD+ and NMN levels. Nat. Metab. 2, 1284–1304 (2020).
Camacho-Pereira, J. et al. CD38 dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. Cell Metab. 23, 1127–1139 (2016).
Bai, P. et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 13, 461–468 (2011).
Pirinen, E. et al. Pharmacological Inhibition of poly(ADP-ribose) polymerases improves fitness and mitochondrial function in skeletal muscle. Cell Metab. 19, 1034–1041 (2014).
Yoshino, J., Mills, K. F., Yoon, M. J. & Imai, S. Nicotinamide mononucleotide, a key NAD+ intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 14, 528–536 (2011).
Cantó, C. et al. The NAD+ precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 15, 838–847 (2012).
Khan, N. A. et al. Effective treatment of mitochondrial myopathy by nicotinamide riboside, a vitamin B3. EMBO Mol. Med. 6, 721–731 (2014).
Cerutti, R. et al. NAD+-dependent activation of Sirt1 corrects the phenotype in a mouse model of mitochondrial disease. Cell Metab. 19, 1042–1049 (2014).
Scheibye-Knudsen, M. et al. A high-fat diet and NAD+ activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metab. 20, 840–855 (2014).
Fang, E. F. et al. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD+/SIRT1 reduction. Cell 157, 882–896 (2014).
Elhassan, Y. S. et al. Nicotinamide riboside augments the aged human skeletal muscle NAD+ metabolome and induces transcriptomic and anti-inflammatory signatures. Cell Rep. 28, 1717–1728.e6 (2019).
Minhas, P. S. et al. Macrophage de novo NAD+ synthesis specifies immune function in aging and inflammation. Nat. Immunol. 20, 50–63 (2019).
Gariani, K. et al. Inhibiting poly ADP-ribosylation increases fatty acid oxidation and protects against fatty liver disease. J. Hepatol. 66, 132–141 (2017).
Katsyuba, E. et al. De novo NAD+ synthesis enhances mitochondrial function and improves health. Nature 563, 354–359 (2018).
Tran, M. T. et al. PGC1α drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 531, 528–532 (2016).
Poyan Mehr, A. et al. De novo NAD+ biosynthetic impairment in acute kidney injury in humans. Nat. Med. 24, 1351–1359 (2018).
Williams, P. A. et al. Vitamin B3 modulates mitochondrial vulnerability and prevents glaucoma in aged mice. Science 355, 756–760 (2017).
Pillai, J. B., Isbatan, A., Imai, S. & Gupta, M. P. Poly(ADP-ribose) polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced Sir2α deacetylase activity. J. Biol. Chem. 280, 43121–43130 (2005).
Yamamoto, T. et al. Nicotinamide mononucleotide, an intermediate of NAD+ synthesis, protects the heart from ischemia and reperfusion. PLoS ONE 9, e98972 (2014).
de Picciotto, N. E. et al. Nicotinamide mononucleotide supplementation reverses vascular dysfunction and oxidative stress with aging in mice. Aging Cell 15, 522–530 (2016).
Hou, Y. et al. NAD+ supplementation normalizes key Alzheimer’s features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc. Natl Acad. Sci. USA 115, E1876–E1885 (2018).
Gong, B. et al. Nicotinamide riboside restores cognition through an upregulation of proliferator-activated receptor-γ coactivator 1α regulated β-secretase 1 degradation and mitochondrial gene expression in Alzheimer’s mouse models. Neurobiol. Aging 34, 1581–1588 (2013).
Altay, O. et al. Combined metabolic activators accelerates recovery in mild-to-moderate COVID-19. Adv. Sci. 8, e2101222 (2021).
Martínez-Reyes, I. & Chandel, N. S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 11, 102 (2020).
Liu, X. et al. The structural basis of protein acetylation by the p300/CBP transcriptional coactivator. Nature 451, 846–850 (2008).
Maksimoska, J., Segura-Peña, D., Cole, P. A. & Marmorstein, R. Structure of the p300 histone acetyltransferase bound to acetyl-coenzyme A and its analogues. Biochemistry 53, 3415–3422 (2014).
Wellen, K. E. et al. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080 (2009).
Lee, J. V. et al. Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab. 20, 306–319 (2014).
Zhao, S. et al. ATP-citrate lyase controls a glucose-to-acetate metabolic switch. Cell Rep. 17, 1037–1052 (2016).
Campbell, S. L. & Wellen, K. E. Metabolic signaling to the nucleus in cancer. Mol. Cell 71, 398–408 (2018).
Martínez-Reyes, I. et al. TCA cycle and mitochondrial membrane potential are necessary for diverse biological functions. Mol. Cell 61, 199–209 (2016).
Nowicki, S. & Gottlieb, E. Oncometabolites: tailoring our genes. FEBS J. 282, 2796–2805 (2015).
Chin, R. M. et al. The metabolite α-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature 510, 397–401 (2014).
Su, Y. et al. α-ketoglutarate extends Drosophila lifespan by inhibiting mTOR and activating AMPK. Aging 11, 4183–4197 (2019).
Edwards, C. B., Copes, N., Brito, A. G., Canfield, J. & Bradshaw, P. C. Malate and fumarate extend lifespan in Caenorhabditis elegans. PLoS ONE 8, e58345 (2013).
Gallo, M., Park, D. & Riddle, D. L. Increased longevity of some C. elegans mitochondrial mutants explained by activation of an alternative energy-producing pathway. Mech. Ageing Dev. 132, 515–518 (2011).
Ishii, N. et al. A mutation in succinate dehydrogenase cytochrome b causes oxidative stress and ageing in nematodes. Nature 394, 694–697 (1998).
Walker, D. W. et al. Hypersensitivity to oxygen and shortened lifespan in a Drosophila mitochondrial complex II mutant. Proc. Natl Acad. Sci. USA 103, 16382–16387 (2006).
Zhu, D. et al. NuRD mediates mitochondrial stress-induced longevity via chromatin remodeling in response to acetyl-CoA level. Sci. Adv. 6, eabb2529 (2020).
Harrison, A. P. & Pierzynowski, S. G. Biological effects of 2-oxoglutarate with particular emphasis on the regulation of protein, mineral and lipid absorption/metabolism, muscle performance, kidney function, bone formation and cancerogenesis, all viewed from a healthy ageing perspective state of the art–review article. J. Physiol. Pharmacol. 59, 91–106 (2008).
Tian, Q. et al. Dietary α-ketoglutarate promotes beige adipogenesis and prevents obesity in middle-aged mice. Aging Cell 19, e13059 (2020).
Fasolino, M., Liu, S., Wang, Y. & Zhou, Z. Distinct cellular and molecular environments support aging-related DNA methylation changes in the substantia nigra. Epigenomics 9, 21–31 (2017).
Wang, Y. et al. α-ketoglutarate ameliorates age-related osteoporosis via regulating histone methylations. Nat. Commun. 11, 5596 (2020).
Niemiec, T. et al. α-ketoglutarate stabilizes redox homeostasis and improves arterial elasticity in aged mice. J. Physiol. Pharmacol. 62, 37–43 (2011).
Asadi Shahmirzadi, A. et al. α-ketoglutarate, an endogenous metabolite, extends lifespan and compresses morbidity in aging mice. Cell Metab. 32, 447–456.e6 (2020).
Zhang, Z. et al. ketoglutarate delays age-related fertility decline in mammals. Aging Cell 20, e13291 (2021).
Carey, B. W., Finley, L. W. S., Cross, J. R., Allis, C. D. & Thompson, C. B. Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 518, 413–416 (2015).
TeSlaa, T. et al. α-ketoglutarate accelerates the initial differentiation of primed human pluripotent stem cells. Cell Metab. 24, 485–493 (2016).
Tischler, J. et al. Metabolic regulation of pluripotency and germ cell fate through α-ketoglutarate. EMBO J. 38, e99518 (2019).
Fogarty, M. J., Marin Mathieu, N., Mantilla, C. B. & Sieck, G. C. Aging reduces succinate dehydrogenase activity in rat type IIx/IIb diaphragm muscle fibers. J. Appl. Physiol. 128, 70–77 (2020).
Bowman, A. & Birch-Machin, M. A. Age-dependent decrease of mitochondrial complex II activity in human skin fibroblasts. J. Invest. Dermatol. 136, 912–919 (2016).
Spinelli, J. B. et al. Fumarate is a terminal electron acceptor in the mammalian electron transport chain. Science 374, 1227–1237 (2021).
Romani, M. et al. NAD+ boosting reduces age-associated amyloidosis and restores mitochondrial homeostasis in muscle. Cell Rep. 34, 108660 (2021).
Benayoun, B. A., Pollina, E. A. & Brunet, A. Epigenetic regulation of ageing: linking environmental inputs to genomic stability. Nat. Rev. Mol. Cell Biol. 16, 593–610 (2015).
Schroeder, E. A., Raimundo, N. & Shadel, G. S. Epigenetic silencing mediates mitochondria stress-induced longevity. Cell Metab. 17, 954–964 (2013).
Tian, Y. et al. Mitochondrial stress induces chromatin reorganization to promote longevity and UPRmt. Cell 165, 1197–1208 (2016).
Merkwirth, C. et al. Two conserved histone demethylases regulate mitochondrial stress-induced longevity. Cell 165, 1209–1223 (2016).
Li, T. Y. et al. The transcriptional coactivator CBP/p300 is an evolutionarily conserved node that promotes longevity in response to mitochondrial stress. Nat. Aging 1, 165–178 (2021).
Yuan, J. et al. Two conserved epigenetic regulators prevent healthy ageing. Nature 579, 118–122 (2020).
Shao, L.-W. et al. Histone deacetylase HDA-1 modulates mitochondrial stress response and longevity. Nat. Commun. 11, 4639 (2020).
Zhang, M. et al. Role of CBP and SATB-1 in aging, dietary restriction, and insulin-like signaling. PLoS Biol. 7, e1000245 (2009).
Haynes, C. M., Yang, Y., Blais, S. P., Neubert, T. A. & Ron, D. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol. Cell 37, 529–540 (2010).
Benedetti, C., Haynes, C. M., Yang, Y., Harding, H. P. & Ron, D. Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics 174, 229–239 (2006).
Haynes, C. M., Petrova, K., Benedetti, C., Yang, Y. & Ron, D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev. Cell 13, 467–480 (2007).
Nargund, A. M., Fiorese, C. J., Pellegrino, M. W., Deng, P. & Haynes, C. M. Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPRmt. Mol. Cell 58, 123–133 (2015).
Acknowledgements
We are grateful to members of the Laboratory of Integrative Systems Physiology (LISP) for helpful comments. This project received funding from the EPFL, the European Research Council under the European Union’s Horizon 2020 research and innovation program (ERC-AdG-787702), the Swiss National Science Foundation (SNSF 31003A_179435), and a GRL grant from the National Research Foundation of Korea (NRF 2017K1A1A2013124). T. Y. L. was supported by the Human Frontier Science Program (LT000731/2018-L). T. L. received support from São Paulo Research Foundation (FAPESP 2019/11171-7).
Author information
Authors and Affiliations
Contributions
T.L. and J.A. conceived the project, drafted the first version, and revised the manuscript. T.Y.L. and A.M. contributed to the conception and writing of the manuscript.
Corresponding author
Ethics declarations
Competing interests
J.A. is a consultant to Mitobridge/Astellas, Amazentis, NOV Metapharma, and Metro Biotech. The other authors have no competing interests.
Peer review
Peer review information
Nature Aging thanks Peter Tessarz, Martin Picard, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Lima, T., Li, T.Y., Mottis, A. et al. Pleiotropic effects of mitochondria in aging. Nat Aging 2, 199–213 (2022). https://doi.org/10.1038/s43587-022-00191-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s43587-022-00191-2
This article is cited by
-
Effect of resistance training plus enriched probiotic supplement on sestrin2, oxidative stress, and mitophagy markers in elderly male Wistar rats
Scientific Reports (2024)
-
Modulation of mitochondrial activity by sugarcane (Saccharum officinarum L.) top extract and its bioactive polyphenols: a comprehensive transcriptomics analysis in C2C12 myotubes and HepG2 hepatocytes
Natural Products and Bioprospecting (2024)
-
Defective mitochondria remodelling in B cells leads to an aged immune response
Nature Communications (2024)
-
Mitophagy curtails cytosolic mtDNA-dependent activation of cGAS/STING inflammation during aging
Nature Communications (2024)
-
Quinolinic acid impairs mitophagy promoting microglia senescence and poor healthspan in C. elegans: a mechanism of impaired aging process
Biology Direct (2023)
