
Abstract
Cellular senescence represents a distinct cell fate characterized by replicative arrest in response to a host of extrinsic and intrinsic stresses. Senescence facilitates programming during development and wound healing, while limiting tumorigenesis. However, pathologic accumulation of senescent cells is implicated in a range of diseases and age-associated morbidities across organ systems. Senescent cells produce distinct paracrine and endocrine signals, causing local tissue dysfunction and exerting deleterious systemic effects. Senescent cell removal by apoptosis-inducing senolytic agents or therapies that inhibit the senescence-associated secretory phenotype have demonstrated benefit in both preclinical and clinical models of geriatric decline and chronic diseases, suggesting that senescent cells represent a pharmacologic target for alleviating effects of fundamental aging processes. However, senescent cell populations are heterogeneous in form, function and tissue distribution, and even differ among species, possibly explaining issues of bench-to-bedside translation in current clinical trials. Here we review features of senescent cells and strategies for targeting them, including immunologic approaches, as well as key intracellular signaling pathways. Additionally, we survey current senolytic therapies in human trials. Collectively, there is demand for research to develop targeted senotherapeutics that address the needs of the aging and chronically ill.
Similar content being viewed by others

Main
Chronological age is the preeminent risk factor for the leading causes of morbidity and mortality worldwide1,2. Chronological and biological aging are often correlated3; however, the latter can be accelerated by the coexistence of multiple chronic diseases and geriatric syndromes, which in turn account for interindividual heterogeneity in terms of risk for development of new diseases, disability or death4. With these considerations in mind, there is now a growing effort to understand and modulate the root causes of aging to alleviate multimorbidity at a global scale. Cellular senescence has garnered increasing attention as a fundamental driver of chronic conditions and functional decline in late life. Despite its historic discovery in the context of in vitro cell growth arrest following serial passage5, the ramifications of cellular senescence have only recently been appreciated following its implication in a host of age-related and chronic diseases spanning most organ systems. In tandem with our expanding appreciation of senescence in pathology, there is an equally evolving understanding of the role of cellular senescence in basic physiology. This inherent duality in the senescence program alongside our limited understanding has increased research fervor and incited controversies ranging from appropriate laboratory methods for studying senescent cells to their potential role as therapeutic targets.
Here we survey the current field of cellular senescence research with regards to its inherent heterogeneity to address the diverse roles of this cell fate with respect to both physiology and pathology. We first explore the molecular underpinnings of senescence to emphasize how senescent cells are currently defined in light of their underlying diversity. We then explore how this heterogeneity is further compounded by the senescent cell secretome. Finally, we consider the therapeutic implications of and approaches for targeting various types of senescent cells as well as the current clinical trials of senolytic drugs.
Cellular senescence
Despite the controversies surrounding the role of cellular senescence in both health and disease, there is a degree of consensus involving its basic definitions and principles. At its core, cellular senescence represents a cell fate that is characterized by stable proliferative arrest in response to various stressors and, frequently, production of an accompanying secretome termed the senescence-associated secretory phenotype (SASP). Inducing stressors vary and include replicative stress with associated telomere shortening, activation of DNA damage pathways, epigenetic changes, oxidative stress, mitochondrial dysfunction, radiation, oncogene induction, and mechanical and shear stress, among others that have been thoroughly reviewed6. Following stress exposure, cell cycle checkpoint blockading factors are upregulated that halt replicative progression. Although these cells accumulate damage products, including DNA breaks, and upregulate DNA damage response (DDR) pathways7,8 in part due to mitochondrial dysfunction and increased reactive oxygen species (ROS) production9, they do not undergo apoptosis. Instead, they have upregulated pro-survival pathways called senescent cell anti-apoptotic pathways (SCAPs) while key apoptotic mediators are downregulated10,11,12,13. In addition to their internal homeostatic perturbations, senescent cells can induce an inflammatory state that provokes both local and systemic inflammation and tissue damage through their SASP14,15,16.
This cascade of stress, senescence, and subsequent SASP signaling and immune activation is critical for normal development and health. From early development, cells with senescent features are important for body axis patterning, with the associated SASP directing growth of structures such as the limb bud and orchestrating regression of transient features through macrophage-mediated clearance17,18,19. Furthermore, as senescent cells produce factors that promote tissue remodeling and recruit associated immune components, spatiotemporally regulated senescence is important for mediating the balance between healing and fibrosis in regenerative processes including wound and organ repair20,21,22,23,24,25. Interestingly, senescent cells can also promote dedifferentiation and plasticity of neighboring cells via paracrine mechanisms26,27. Additionally, owing to its induction in response to both DNA damage and oncogenic stress, the senescence program may also serve as a tumor suppression mechanism, although its inflammatory properties and DNA damage induction are also associated with increased tumor burden when dysregulated28,29,30.
While there is substantial interest in the beneficial roles of senescent cells, much of cellular senescence research has been aimed at their roles in disease and emphasized their contributions to geriatric decline. This modern wave of attention was promoted by the discoveries that markers of cellular senescence accumulate with aging and accumulation is delayed by interventions that increase healthspan and lifespan31, removal of senescent cells increases healthspan in progeroid mouse models32, and transplantation of a relatively small number of senescent cells into previously healthy animals provokes multisystem dysfunction similar to what is seen in aged animals33,34. Furthermore, it was found that the correlation of senescent cell accumulation with disease extends to humans35,36,37, and that senescent cell burden can be safely reduced in a clinical context38. On the basis of these points, it appears crucial to understand the roles of senescence in morbidity and mortality. To date, deleterious effects of senescent cells have been implicated in a causative or proximal capacity for diseases encompassing most organ systems, including the leading causes of morbidity, death and healthcare burden, such as cardiovascular, pulmonary, neurologic, renal, hepatic, infectious, musculoskeletal and endocrine diseases29,39,40,41,42,43,44,45,46,47,48,49,50,51.
Molecular basis and markers of cellular senescence
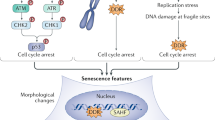
Senescence is a cell fate that is widely regarded as a stress response. Senescence is distinct from other non-proliferative cell fates including quiescence, the latter being reversible with appropriate mitogens, or terminal differentiation, in which cells develop from defined lineages to fulfill specific roles and arrest growth through diverse pathways. In response to internal or external factors such as telomere shortening or ionizing radiation, senescent cell cycle arrest signals primarily converge on the p53–Cdkn1a (p21) and/or retinoblastoma (RB)–Cdkn2a (p16) pathways52. Increased levels of the cyclin-dependent kinase inhibitors (CDKIs) p21 or p16 can contribute to G1/S cell cycle blockade, mediated by inhibition of the formation of the cyclin–cyclin-dependent kinase complex and failure to inactivate RB with associated transcriptional repression53. Despite the convergence of senescent growth arrest pathways, there is growing appreciation that different inducing stressors can result in unique senescent subpopulations54. This complexity is further compounded by the fact that currently no single known marker defines senescence with high sensitivity and specificity, although this lack of a gold standard is not unique to the field. Current practices rely on a combination of phenotypes and sets of markers to suggest cellular senescence. For example, although accumulation of p16 and p21 has been biologically implicated in senescence and they are extensively used as molecular markers, various in vitro stressors induce differential expression of these CDKIs, even within the same cell line55, indicating that neither is fully sufficient or reliable on its own. To complicate this further, the same study also revealed that different cell types can differentially alter their transcriptome in response to the same inducing stressor. Unlike the well-controlled constraints of cell culture models, different stressors can act simultaneously in vivo with both aging and disease. Thus, it is hypothesized that senescent populations are especially diverse in vivo.
Besides p16 and p21, a number of other markers can be used to identify senescent cells, although sensitivity and specificity varies for each (Table 1). Several morphologic features are suggestive of senescence in vitro and can be assessed using approaches such as brightfield microscopy. Compared to their counterparts, senescent cells have increased size and granularity56,57, likely reflecting their altered metabolism and organelle homeostasis. In particular, both mitochondria and lysosomes accumulate in cells exposed to senescence-inducing stressors9,58. Whether causative of, or occurring in response to the stress response, or both, these changes provide an added means for detecting senescent cells. Lysosomal abundance in senescent cells is one of the most commonly used features, as lysosomal senescence-associated beta-galactosidase (SA-β-gal) activity assays are relatively quick and straightforward to conduct59. However, there are limitations to the specificity of this marker60 and it should be paired with others considered here. Lipofuscins, which are aggregates of lysosomal byproducts, also accumulate within senescent cells and can conveniently be assayed, although such assays have similar limitations to specificity to those of SA-β-gal assays61. Mitochondrial abundance increases in senescent cells together with altered membrane potential and increased ROS production, oxidative phosphorylation and oxygen consumption62. Hence, assays of redox state, mitochondrial function and mitochondrial biogenesis can be used to enhance senescent cell phenotyping.
Nuclear alterations are prominent features of senescent cells and largely result from activation of the DDR pathways that lead to p16 and p21 activation. These pathways also lead to increased phosphorylation and activation of the DDR orchestrator histone, H2AX, which coordinates DNA repair following genotoxic stress and double-strand breaks63. This has led to the use of immunohistochemical techniques to label phosphorylated (γ) H2AX in senescence assays. As this pathway can repair DNA double-strand breaks across most of the chromosome, it potentially lacks sensitivity owing to its transient nature at most loci. However, double-strand breaks are persistent at telomeric loci owing to localized inhibition of non-homologous end-joining pathways developed to prevent chromosomal end-to-end fusions64. As a result, assays for telomere-associated foci of DNA damage, assessed by assaying for colocalization of telomeric probes and γ-H2AX antibodies, which accumulate in cells following senescence-inducing stress, are relatively sensitive and specific for detecting senescent cells8. Other nuclear alterations include loss of the nuclear intermediate filament protein and the epigenetic modulator, lamin B1 (ref. 65), specific chromatin remodeling patterns termed senescence-associated heterochromatin foci66 and decreased DNA replication, each of which can be assessed using immunofluorescence techniques. Additional genetic and epigenetic markers include de-repression of LINE-1 retrotransposons67 and upregulation of the histone acetyltransferase KAT7 (ref. 68).
The cellular senescence research field is currently being propelled by omic and bioinformatic approaches that may elicit a better understanding of the senescence landscape and provide new molecular insights with a particular emphasis on heterogeneity. Transcriptomic approaches yield unique perspectives into the dynamics of senescent cells while suggesting novel markers. It was recently revealed that, regardless of the inducing stress and cell type, there appear to be shared profiles of differential gene expression in senescent cells55. This includes a shared downregulation of histone-associated transcripts and upregulation of the long noncoding RNA PURPL that is involved in p53 signal modulation. However, at single-cell resolution, transcriptomic analysis reveals that even a single cell faced with identical stressors yields unique senescent subtypes with distinct profiles of differential gene and SASP expression69, calling into question the possibility of ever finding a single gold standard senescence marker. Regardless, single-cell transcriptomics allow naturally occurring senescent cell populations to be examined by leveraging a multitude of marker transcripts. For example, this technique has been used to show that adipose-derived mesenchymal stem cells accumulate senescence markers with natural aging and altered senescence-associated pathways70. Coupled with epigenomic insight71, and proteomic72 and metabolomic73 data, our understanding of the senescent cell fate is growing exponentially, and we are on the path to a comprehensive profiling database.
The SASP
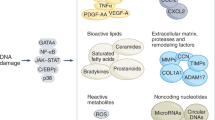
In addition to the effects mediated by their proliferative arrest, the SASP is a key contributor to the combined physiologic and pathologic roles of senescent cells. The SASP represents a myriad of cytokines, matrix metalloproteinases, microRNAs, chemokines, growth factors and small-molecule metabolites that are directly secreted or exosomally packaged6,15,74 and are largely under the control of the p53, NF-κB, CEBPB, JAK–STAT and GATA4 transcription factors75,76,77,78. Beyond the transcriptional level, there is an evolving understanding that SASP regulation is multilayered, including contributions from pre-transcriptional signaling cascades such as the cGAS–STING pathway79,80,81 as well as epigenetic regulation82,83 with particular remodeling noted in the super-enhancer landscape84. Efforts are being made, such as by the SASP Atlas, to map and record the resulting senescence secretome, and such analyses already indicate that the SASP varies with different stressors72. For example, pathogen-related factors, such as lipopolysaccharide or severe acute respiratory syndrome coronavirus 2 S1 antigen, can substantially amplify the SASP of existing senescent cells, potentially contributing to the increased risks for cytokine storm and mortality in the elderly and those with chronic diseases that are associated with a high burden of senescent cells due to infections50.
SASP components fulfill many functions, but are classically associated with the chronic inflammatory state of aging85,86. Several SASP components are immunomodulatory87. SASP chemokines such as interleukin 1 (IL-1), IL-8 and tumor necrosis factor (TNF) recruit immune cells including macrophages, neutrophils and T cells88,89,90. This allows for senescent and neighboring cells that have accumulated ROS, DNA damage or oncogenic stress to be destroyed, consistent with the protective role of senescence in tumor prevention. Conversely, these same SASP components (alongside others) when chronically or uncontrollably expressed may perpetuate tumor seeding and invasion through matrix and vascular remodeling. This could explain the apparently contradictory findings implicating senescence in both cancer prevention and progression91,92,93,94. Furthermore, the SASP is not only a product of senescent cells, it also induces and reinforces senescence itself through paracrine and autocrine signaling mechanisms34,95,96. In light of this, it is thought that age-associated losses in immune competency in a chronically inflamed milieu may allow senescent cells to evade removal97,98,99, although this is not fully understood. Additionally, SASP signals confer senescent cells with resistance to immune-mediated clearance (for example, through upregulation of ligands such as HLA-E, which can inhibit CD8+ T cell and natural killer cell function100). Regardless of the exact underlying mechanism of immune interplay, senescent cell accumulation can perpetuate a feed-forward cycle of failed clearance coupled with increased SASP signals and resulting secondary senescent cell induction (Fig. 1). Of note, though, there is debate over whether cells that are secondarily induced into senescence are similar to those becoming senescent owing to a primary stressor, with evidence indicating differences in SASP dynamics between primarily and secondarily induced senescent cells101. Additionally, senescence and damaging SASP signals are further augmented by aged immune system components. For example, T cells with mitochondrial dysfunction, like those present with aging, cause global increases in senescent cell burden that are associated with increases in frailty and multimorbidity and that appear to be partially mediated by TNF signaling102. Furthermore, introducing a construct that interferes with DNA repair into hematopoietic progenitors in transgenic mice, so that immune cells prematurely become senescent, causes early development of multiple age-related phenotypes and increases senescent cell burden in multiple tissues in transgenic mice103.

Cellular senescence is induced following exposure to an initial stress. The resulting senescent cells produce a SASP that can potentiate further senescent cell accumulation and impair clearance. In turn, the SASP can be amplified, eliciting an environment of chronic inflammation and additional senescence-inducing stressors that drive a feed-forward cycle of senescent cell accumulation.
SASP heterogeneity, reflecting the complexity of the underlying senescence program, is further recapitulated in how the SASP impacts the regenerative potential of non-senescent cells. When expressed transiently, NF-κB pathway SASP factors can promote stemness and enhance regenerative potential in keratinocyte regeneration models in vivo27. However, interruption of this signal or prolonged exposure to it reduces stemness markers and impairs regenerative capacity27. This highlights the need to consider SASP kinetics and illustrates how chronic senescent cell accumulation and signaling can impair ability to respond to physiologic insults. Opposing roles of the senescent secretome are also evident in the liver. Senescence and associated transforming growth factor-β signaling have been linked with impaired regeneration and biliary fibrosis104, while genetic ablation of senescent cells and ensuing SASP reduction can induce fibrosis in vascular structures105. Although these findings may be artifacts of the underlying model systems used, including continuous elimination of senescent cells, limited specificity of p16 as a senescence marker, and potentially vascular cell populations with associated vascular leakage in the latter study105, there appears to be contextual variability in features of senescence.
Targeting senescent cells
A 2004 report31 of an inverse relation between senescent cell burden and healthspan in mice prompted the initiation of efforts to develop drugs that selectively eliminate senescent cells. The subsequent development of the transgenic INK-ATTAC mouse model32, in which age-related phenotypes were alleviated through selectively removing cells expressing p16 at high levels from progeroid mice, further supported these efforts. As senescent cells are highly heterogeneous in both their molecular biology and their physiological function, targeted strategies are needed that ideally preserve senescent cells in beneficial contexts while eliminating effects that are detrimental. Broadly, these therapies can be broken down into the major categories of senomorphic and senolytic drugs, although this classification might be arbitrary as agents with senomorphic effects in one cell type or context may be senolytic in another and vice versa. Senomorphic compounds target pathologic SASP signaling, while senolytics eliminate the underlying senescent cells that release damaging SASP factors. Senomorphics are discussed elsewhere, but in brief senomorphics prevent the production of, antagonize or neutralize SASP components106,107, and usually require continuous administration. We will instead focus on emerging senolytic strategies that address a root cause of senescence pathology, senescent cells, yielding pleiotropic benefits with intermittent administration. The current senolytic class originates from work demonstrating senescent cell clearance with the combination of the Src kinase inhibitor dasatinib and the flavonoid quercetin (D + Q)10 and, subsequently, BCL-2 family inhibitors and others108,109,110,111,112. While the first senolytics were developed using a bioinformatically informed approach aimed at disrupting SCAPs and other pro-survival networks10, the class has expanded to take advantage of additional senescence features and enhance immune-mediated clearance (Fig. 2). Broadly, first-generation agents act by transiently disabling SCAPs, causing those senescent cells with a tissue-damaging SASP to kill themselves. Importantly, while all senolytic strategies may elicit off-target effects or interfere with beneficial populations, these can often be limited as most therapeutics are amenable to intermittent ‘hit-and-run’ dosing strategies that do not require daily, or even weekly, administration. This interval dosing strategy is hypothesized to be efficacious given that senescent cells take 7 days or more (at least in vitro) to accumulate and develop a SASP even when faced with persistent and harsh senescence-inducing stress in vitro67,113, and could potentially take as long to start re-accumulating in vivo.

Select features of senescent cells have been leveraged to specifically reduce their abundance. Broadly, these targets include unique surface markers, SCAP and other survival networks, biochemical adaptations and changes in organelle characteristics.
Extracellular targets and immune-mediated clearance
Characterization of senescent cells has revealed unique markers that serve as senescence-associated self-antigens. These can be co-opted for immune system–mediated senolytic activity and clearance. A recent study114 took advantage of this using chimeric antigen receptor (CAR) T cells targeted against the urokinase-type plasminogen activator receptor (uPAR) in a mouse model. uPAR is associated with extracellular matrix remodeling that is upregulated at the cell surface of senescent cells during replicative, oncogene-induced and toxicity-induced senescence. Cytotoxic CAR T cells were able to selectively clear uPAR-expressing senescent cells in vitro and in vivo. CAR T cell–mediated clearance of senescent cells led to survival and histopathologic benefit in murine models of both carbon tetrachloride– and diet-induced liver fibrosis, suggesting the feasibility and potential of this clearance strategy. However, supratherapeutic CAR T cell dosing is associated with a proinflammatory cytokine profile, weight loss and hypothermia, suggesting that careful attention would need to be paid to initial dosing strategies, particularly in geriatric populations where a compromised immune system together with ongoing chronic inflammation may restrict the therapeutic window. Additionally, CAR T cell approaches in humans involve initial immune system suppression with considerable attendant morbidity and are expensive. Although senolytic CAR T cells appear to be self-limiting114, their expansion and contraction occurs over a period of days, which is a risk in the case of an acute to subacute adverse reaction. Overall, this strategy is highly promising given its preclinical efficacy and senescent cell specificity, but future optimization and testing is necessary before clinical administration.
Co-opted senescence antigens provide an additional strategy for cell clearance through vaccination. Senescent T cell populations accumulate in obese adipose tissue, contributing to inflammation with both local and systemic effects115. These T cell populations bear known senescence-associated markers and are characterized by CD4+, CD44high, CD62Llow, PD1+ and CD153+ expression. Immunization against these cells with a CD153 peptide conjugate promotes senescent T cell clearance from adipose tissue with associated improvements in metabolic function. As with other immune-mediated clearance strategies, vaccination against senescence epitopes will require careful consideration for clinical translation especially in the context of their lasting effects that might be difficult to reverse. As CD153 is implicated in mycobacterial clearance116, neutralizing antibodies or elimination of positive cells may be contraindicated in those with a history of or enhanced susceptibility to mycobacterial infection. Additionally, immunosenescence may limit vaccine response and memory in the geriatric population117.
Modulating SCAPs
Prototypic senolytic drugs were developed to target SCAP networks10. In contrast to a one-drug, one-target approach, SCAP inhibition may interface with several pro-survival signals at once. As a result, these early senolytics typically possess several pharmacologic mechanisms of action that interact synergistically. Prime examples of this are the flavonoid fisetin118 as well as D + Q, which have been utilized and reviewed thoroughly112. In brief, although the precise mechanism of action is unknown (as is the case for most agents), the D + Q combination exerts broad spectrum senolytic activity through interference with several pro-survival networks, including ephrin dependence receptor signaling, PI3K–AKT and BCL-2 members. The BCL-2 family members (BCL-2, BCL-XL, BCL-W and so on), which prevent activation of pro-apoptotic mitochondrial signaling cascades, cytochrome c release and downstream caspase activation, are targeted by other senolytics. These compounds, such as navitoclax (ABT-263), A1331852 and A1155463, display in vitro activity against senescent human lung fibroblasts and umbilical vein endothelial cells10,119 amongst other cell types120. However, in contrast to D + Q, these drugs do not eliminate all senescent populations in vitro, with particular resilience demonstrated by senescent pre-adipocytes that are capable of causing widespread metabolic dysfunction and harm in vivo34. Additionally, the clinical utility of BCL-2 inhibitors as senolytics is limited by off-target effects on platelets121, neutrophils122 and potentially T cells123, which can be idiosyncratic (that is, can occur after a single dose) and might impair hemostasis and further compromise immune function in at-risk populations, although intermittent dosing may help. Additional SCAPs that can be drug targeted include modulation of p53-associated pathways and interruption of the anti-apoptotic transcription factor FOXO4 (ref. 12), inhibition of the peptidase USP7 (ref. 124), inhibition of the HSP90 chaperones125, increased apoptotic drive by caspase activation126 and others that are being studied.
Other approaches
Additional aspects of senescent cells are advantageous for directed senolysis. One such feature is their increased lysosomal enzyme activity. This can be leveraged with the use of prodrugs that are cleaved and activated by the lysosomal enzyme SA-β-gal or by loading cytotoxic chemicals into galacto-polymer-coated nanoparticles that can be preferentially released into senescent cells127,128. This strategy could suffer from off-target effects in non-senescent cells with high SA-β-gal activity, such as activated macrophages129. Lysosomal abundance in senescent cells also offers the opportunity for senolysis through increased autophagy. Autophagy is normally involved with the turnover of organelles and other cellular components that are directed to the lysosome for molecular repurposing. Outside its role as a cellular recycling system, autophagy can lead to activation of cell death pathways when highly activated under persistent stress130. Autophagy is inhibited within senescent cells131, but senescent cells are primed for cell death following an autophagic push. This is demonstrated by autophagy induction and subsequent senolysis with the use of fibrate drugs132, metformin133, mTOR complex 1 (mTORC1) inhibitors134, bromodomain and extraterminal domain (BET) inhibition135 and lysosomal acidification with ataxia telangiectasia mutated (ATM) inhibitors136. Beyond organelle targeting, even minor chemical changes can be exploited for preferential senolysis. This is demonstrated by the senolytic activity of cardiac glycosides, which take advantage of changes in membrane potentials and proton concentrations within senescent cells137,138. Furthermore, compensatory responses to these chemical changes in senescent cells may be targeted. For example, senescent cells resist lowered pH produced by lysosomal alterations and other factors by upregulating buffering systems including the glutaminase product ammonia139. Interruption of these buffering systems can induce preferential senescent cell death, and the enzymes that maintain buffer concentrations could be targeted by small-molecule drugs.
Clinical trials of senolytics
The first clinical study of senolytics published was a pilot, open-label study of 14 patients with idiopathic pulmonary fibrosis140 (ClinicalTrials.gov identifier NCT02874989). They were administered 9 doses of oral D + Q over 3 weeks. Five days after the final dose, the participants had improved 6-minute walk distance, walking speed, chair rise ability and short physical performance battery. This may have been related to the study drug, learning effects or other factors inherent in open-label study designs. On the basis of these initial data, a larger, placebo-controlled clinical trial of D + Q for idiopathic pulmonary fibrosis is planned. Interim results were recently reported from a phase 1, open-label, clinical trial of D + Q in individuals with diabetic kidney disease (NCT02848131). By 11 days after the last day of a 3-day oral course of D + Q, 9 individuals with diabetic kidney disease had decreased numbers of adipose tissue p16INK4A-positive and SA-β-gal+ cells compared to those in biopsies before D + Q was given38,141. Additionally, 11 days after completing the 3-day senolytic intervention, a composite score of 10 circulating SASP factors was significantly decreased as was activated CD68+ macrophage adipose tissue infiltration and adipose tissue crown-like structures, which are due to fibrosis. In this continuing trial (goal = 30 individuals), no serious or severe side-effects have emerged so far. Multiple other clinical trials are underway or about to begin (Table 2). These include, among others, trials of D + Q for Alzheimer’s disease (ALSENLITE, NCT04785300; SToMP-AD, NCT04685590), the accelerated aging-like state in bone marrow transplant survivors (HTSS, NCT02652052), the accelerated aging-like state in survivors of childhood cancer (SENSURV, NCT04733534) and age-related osteoporosis (NCT04313634), and trials of fisetin for frailty in older women (AFFIRM, NCT03430037), diabetic and chronic kidney disease (NCT03325322), childhood cancer survivors (compared to D + Q in SENSURV), age-related osteoporosis (compared to D + Q in NCT04313634), osteoarthritis (NCT04210986) and coronavirus in individuals in nursing homes, hospitalized individuals and outpatients (COVID-FIS, NCT04537299; COVID-FISETIN, NCT04476953; and COVFIS-HOME, NCT04771611, respectively). In addition to D + Q and fisetin, a trial of the Bcl-xL inhibitor UBX1325 for the treatment of diabetic macular edema is currently recruiting (NCT04537884). A clinical trial of the proposed senolytic compound UBX0101, an inhibitor of the murine double minute 2 (MDM2) negative regulator of p53, for treatment of osteoarthritis (NCT03513016) was recently halted in phase 2 following failure to outperform the placebo in interim analysis. This is speculated to be due to several factors including that MDM2 inhibitors have senomorphic activity, which could represent their primary mechanism of action and would require continuous administration142. Additionally, UBX0101 required a treatment course of multiple injections in preclinical models143 in contrast to the single injection in the clinical trial. Potential benefits of the UBX0101 arm of the trial may also have been masked due to the clinical benefits of intra-articular saline injection on patient-reported outcomes in osteoarthritis144. Until results and data about benefits as well as adverse or off-target events are available from these and other studies, senolytic agents should not be administered outside carefully monitored clinical trials.
Conclusions and future directions
In the last decade there has been exponential growth in cellular senescence research due to its pervasive and widespread implications. Additional field-defining discoveries are certain to follow over the coming years. However, substantial gaps and key questions still remain. Senescent cells exhibit hysteresis, in that their phenotypes are dependent on the context of prior stress. Furthermore, senescent cells are heterogeneous in both form and function. Senescent phenotypes vary by cell type, tissue of origin, tissue of residence, functional impact and how senescence was induced (Fig. 3). This is further complicated by differences between in vitro models and translation from tissue cultures studies to in vivo animal models and ultimately to humans. However, few models exist to study these differences (Table 3). Current animal models such as INK-ATTAC (ref. 32) or p16-3MR mice25 and other p16-dependent systems105,145,146,147 are limited because p16Ink4a is not expressed by every senescent cell and non-senescent, frequently abundant cell types, such as activated macrophages, can express p16Ink4a at high levels129. Murine systems using p21-based constructs have recently been developed to address these issues and explore uncharacterized senescent cell populations148. However, it is ultimately unknown whether senescent cells in mice are representative of those found in humans. We have begun to address this through transplantation studies34, in which human senescent cells are introduced into murine hosts, but this question remains open. The field is in clear need of novel animal models, senescent cell culture systems and additional human translational experiments to push forward. Amid this evolving appreciation of senescent cell heterogeneity, it is likely that there are yet undefined populations and subpopulations of senescent cells that may have distinct phenotypes, possess unique spatiotemporal dynamics and fulfill distinct physiologic and pathogenic roles in a context-dependent manner. In particular, few studies have been conducted to assess the characteristics of naturally occurring senescent cells70, which are likely to be the most relevant to aging phenotypes. While there is an abundance of preclinical evidence indicating that senolytics may treat various disease processes or even supplement regenerative therapies149, it is unknown how modulating different senescent cell populations with senotherapeutic strategies will translate into clinical outcomes or whether senolytics differ in efficacy when used preventatively or after a disease process has already begun. Ideally, as our understanding of senescence heterogeneity expands, it will be increasingly possible to use rational design strategies to target and eliminate only the most detrimental senescent cell subpopulations (for example, by leveraging single-cell differential gene expression profiles across senescent cells). However, these translational questions are challenging to address considering the inherent variations between animal models and humans, which are convoluted by differences in immune function and kinetics of senescent cell accumulation. Collectively, these gaps need to be addressed by the burgeoning cellular senescence field and are likely to evoke interest for the foreseeable future.

Senescent cells exhibit context-dependent phenotypic diversity. Numerous stimuli can induce a stress response sufficient to induce cellular senescence. Furthermore, susceptibility to these stressors and senescent cell characteristics can depend on cell type, tissue, disease state and other context. The resulting senescent cells display a spectrum of convergent and divergent phenotypes in their secretome and consequent functionality.
References
Miller, R. A. Extending life: scientific prospects and political obstacles. Milbank Q. 80, 155–174 (2002).
Niccoli, T. & Partridge, L. Ageing as a risk factor for disease. Curr. Biol. 22, R741–R752 (2012).
Li, X. et al. Longitudinal trajectories, correlations and mortality associations of nine biological ages across 20-years follow-up. Elife https://doi.org/10.7554/eLife.51507 (2020).
Ferrucci, L. & Kuchel, G. A. Heterogeneity of aging: individual risk factors, mechanisms, patient priorities, and outcomes. J. Am. Geriatr. Soc. https://doi.org/10.1111/jgs.17011 (2021).
Hayflick, L. & Moorhead, P. S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 25, 585–621 (1961).
Gorgoulis, V. et al. Cellular senescence: defining a path forward. Cell 179, 813–827 (2019).
d’Adda di Fagagna, F. et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature 426, 194–198 (2003).
Hewitt, G. et al. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 3, 708 (2012).
Passos, J. F. et al. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 5, e110 (2007).
Zhu, Y. et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 14, 644–658 (2015).
Kirkland, J. L. & Tchkonia, T. Cellular senescence: a translational perspective. EBioMedicine 21, 21–28 (2017).
Baar, M. P. et al. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell 169, 132–147 (2017).
Marcotte, R., Lacelle, C. & Wang, E. Senescent fibroblasts resist apoptosis by downregulating caspase-3. Mech. Ageing Dev. 125, 777–783 (2004).
Schnabl, B., Purbeck, C. A., Choi, Y. H., Hagedorn, C. H. & Brenner, D. Replicative senescence of activated human hepatic stellate cells is accompanied by a pronounced inflammatory but less fibrogenic phenotype. Hepatology 37, 653–664 (2003).
Coppe, J. P. et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 6, 2853–2868 (2008).
Hari, P. et al. The innate immune sensor Toll-like receptor 2 controls the senescence-associated secretory phenotype. Sci. Adv. 5, eaaw0254 (2019).
Storer, M. et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 155, 1119–1130 (2013).
Davaapil, H., Brockes, J. P. & Yun, M. H. Conserved and novel functions of programmed cellular senescence during vertebrate development. Development 144, 106–114 (2017).
Lorda-Diez, C. I. et al. Apoptosis during embryonic tissue remodeling is accompanied by cell senescence. Aging (Albany NY) 7, 974–985 (2015).
Da Silva-Alvarez, S. et al. Cell senescence contributes to tissue regeneration in zebrafish. Aging Cell 19, e13052 (2020).
Meyer, K., Hodwin, B., Ramanujam, D., Engelhardt, S. & Sarikas, A. Essential role for premature senescence of myofibroblasts in myocardial fibrosis. J. Am. Coll. Cardiol. 67, 2018–2028 (2016).
Feng, T. et al. CCN1-induced cellular senescence promotes heart regeneration. Circulation 139, 2495–2498 (2019).
Jun, J. I. & Lau, L. F. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 12, 676–685 (2010).
Krizhanovsky, V. et al. Senescence of activated stellate cells limits liver fibrosis. Cell 134, 657–667 (2008).
Demaria, M. et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 31, 722–733 (2014).
Mosteiro, L. et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science https://doi.org/10.1126/science.aaf4445 (2016).
Ritschka, B. et al. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. 31, 172–183 (2017).
Schosserer, M., Grillari, J. & Breitenbach, M. The dual role of cellular senescence in developing tumors and their response to cancer therapy. Front. Oncol. 7, 278 (2017).
Prieto, L. I. & Baker, D. J. Cellular senescence and the immune system in cancer. Gerontology 65, 505–512 (2019).
Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 75, 685–705 (2013).
Krishnamurthy, J. et al. Ink4a/Arf expression is a biomarker of aging. J. Clin. Invest. 114, 1299–1307 (2004).
Baker, D. J. et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479, 232–236 (2011).
Xu, M. et al. Transplanted senescent cells induce an osteoarthritis-like condition in mice. J. Gerontol. A Biol. Sci. Med. Sci. 72, 780–785 (2017).
Xu, M. et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 24, 1246–1256 (2018).
Musi, N. et al. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell 17, e12840 (2018).
Justice, J. N. et al. Cellular senescence biomarker p16INK4a+ cell burden in thigh adipose is associated with poor physical function in older women. J. Gerontol. A Biol. Sci. Med. Sci. 73, 939–945 (2018).
Hickson, L. J. et al. Diabetic kidney disease alters the transcriptome and function of human adipose-derived mesenchymal stromal cells but maintains immunomodulatory and paracrine activities important for renal repair. Diabetes https://doi.org/10.2337/db19-1268 (2021).
Hickson, L. J. et al. Senolytics decrease senescent cells in humans: preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 47, 446–456 (2019).
Martinez-Cue, C. & Rueda, N. Cellular senescence in neurodegenerative diseases. Front. Cell Neurosci. 14, 16 (2020).
Shimizu, I. & Minamino, T. Cellular senescence in cardiac diseases. J. Cardiol. 74, 313–319 (2019).
Hansel, C., Jendrossek, V. & Klein, D. Cellular senescence in the lung: the central role of senescent epithelial cells. Int. J. Mol. Sci. https://doi.org/10.3390/ijms21093279 (2020).
Frey, N., Venturelli, S., Zender, L. & Bitzer, M. Cellular senescence in gastrointestinal diseases: from pathogenesis to therapeutics. Nat. Rev. Gastroenterol. Hepatol. 15, 81–95 (2018).
Schmid, N. et al. Insights into replicative senescence of human testicular peritubular cells. Sci. Rep. 9, 15052 (2019).
Guo, M. Cellular senescence and liver disease: mechanisms and therapeutic strategies. Biomed. Pharmacother. 96, 1527–1537 (2017).
Baar, M. P., Perdiguero, E., Munoz-Canoves, P. & de Keizer, P. L. Musculoskeletal senescence: a moving target ready to be eliminated. Curr. Opin. Pharmacol. 40, 147–155 (2018).
Farr, J. N. & Khosla, S. Cellular senescence in bone. Bone 121, 121–133 (2019).
Docherty, M. H., O’Sullivan, E. D., Bonventre, J. V. & Ferenbach, D. A. Cellular senescence in the kidney. J. Am. Soc. Nephrol. 30, 726–736 (2019).
Gruber, F., Kremslehner, C., Eckhart, L. & Tschachler, E. Cell aging and cellular senescence in skin aging - recent advances in fibroblast and keratinocyte biology. Exp. Gerontol. 130, 110780 (2020).
Khosla, S., Farr, J. N., Tchkonia, T. & Kirkland, J. L. The role of cellular senescence in ageing and endocrine disease. Nat. Rev. Endocrinol. 16, 263–275 (2020).
Camell, C. D. et al. Senolytics reduce coronavirus-related mortality in old mice. Science https://doi.org/10.1126/science.abe4832 (2021).
Zhou, Y. et al. Senolytics alleviate the degenerative disorders of temporomandibular joint in old age. Aging Cell https://doi.org/10.1111/acel.13394 (2021).
Martinez-Zamudio, R. I., Robinson, L., Roux, P. F. & Bischof, O. SnapShot: cellular senescence pathways. Cell 170, 816 (2017).
Giacinti, C. & Giordano, A. RB and cell cycle progression. Oncogene 25, 5220–5227 (2006).
Hernandez-Segura, A. et al. Unmasking transcriptional heterogeneity in senescent cells. Curr. Biol. 27, 2652–2660 (2017).
Casella, G. et al. Transcriptome signature of cellular senescence. Nucleic Acids Res. 47, 11476 (2019).
Biran, A. et al. Quantitative identification of senescent cells in aging and disease. Aging Cell 16, 661–671 (2017).
Gosselin, K. et al. Senescent keratinocytes die by autophagic programmed cell death. Am. J. Pathol. 174, 423–435 (2009).
Robbins, E., Levine, E. M. & Eagle, H. Morphologic changes accompanying senescence of cultured human diploid cells. J. Exp. Med. 131, 1211–1222 (1970).
Debacq-Chainiaux, F., Erusalimsky, J. D., Campisi, J. & Toussaint, O. Protocols to detect senescence-associated beta-galactosidase (SA-βgal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc. 4, 1798–1806 (2009).
Yang, N. C. & Hu, M. L. The limitations and validities of senescence associated-beta-galactosidase activity as an aging marker for human foreskin fibroblast Hs68 cells. Exp. Gerontol. 40, 813–819 (2005).
Salmonowicz, H. & Passos, J. F. Detecting senescence: a new method for an old pigment. Aging Cell 16, 432–434 (2017).
Summer, R. et al. Activation of the mTORC1/PGC-1 axis promotes mitochondrial biogenesis and induces cellular senescence in the lung epithelium. Am. J. Physiol. Lung Cell. Mol. Physiol. 316, L1049–L1060 (2019).
Bonner, W. M. et al. GammaH2AX and cancer. Nat. Rev. Cancer 8, 957–967 (2008).
Smogorzewska, A., Karlseder, J., Holtgreve-Grez, H., Jauch, A. & de Lange, T. DNA ligase IV-dependent NHEJ of deprotected mammalian telomeres in G1 and G2. Curr. Biol. 12, 1635–1644 (2002).
Freund, A., Laberge, R. M., Demaria, M. & Campisi, J. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell 23, 2066–2075 (2012).
Aird, K. M. & Zhang, R. Detection of senescence-associated heterochromatin foci (SAHF). Methods Mol. Biol. 965, 185–196 (2013).
De Cecco, M. et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 566, 73–78 (2019).
Wang, W. et al. A genome-wide CRISPR-based screen identifies KAT7 as a driver of cellular senescence. Sci. Transl. Med. https://doi.org/10.1126/scitranslmed.abd2655 (2021).
Chen, W. et al. Single-cell transcriptome analysis reveals six subpopulations reflecting distinct cellular fates in senescent mouse embryonic fibroblasts. Front. Genet. 11, 867 (2020).
Wang, B. et al. Transplanting cells from old but not young donors causes physical dysfunction in older recipients. Aging Cell 19, e13106 (2020).
Yang, N. & Sen, P. The senescent cell epigenome. Aging (Albany NY) 10, 3590–3609 (2018).
Basisty, N. et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 18, e3000599 (2020).
Fernandez-Rebollo, E. et al. Senescence-associated metabolomic phenotype in primary and iPSC-derived mesenchymal stromal cells. Stem Cell Rep. 14, 201–209 (2020).
Borghesan, M. et al. Small extracellular vesicles are key regulators of non-cell autonomous intercellular communication in senescence via the interferon protein IFITM3. Cell Rep. 27, 3956–3971 (2019).
Kang, C. et al. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 349, aaa5612 (2015).
Salminen, A., Kauppinen, A. & Kaarniranta, K. Emerging role of NF-κB signaling in the induction of senescence-associated secretory phenotype (SASP). Cell Signal. 24, 835–845 (2012).
Huggins, C. J. et al. C/EBPγ suppresses senescence and inflammatory gene expression by heterodimerizing with C/EBPβ. Mol. Cell. Biol. 33, 3242–3258 (2013).
Xu, M. et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc. Natl Acad. Sci. USA 112, E6301–E6310 (2015).
Yang, H., Wang, H., Ren, J., Chen, Q. & Chen, Z. J. cGAS is essential for cellular senescence. Proc. Natl Acad. Sci. USA 114, E4612–E4620 (2017).
Li, T. & Chen, Z. J. The cGAS–cGAMP–STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 215, 1287–1299 (2018).
Gluck, S. et al. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol. 19, 1061–1070 (2017).
Hayakawa, T. et al. SIRT1 suppresses the senescence-associated secretory phenotype through epigenetic gene regulation. PLoS ONE 10, e0116480 (2015).
Nacarelli, T., Liu, P. & Zhang, R. Epigenetic basis of cellular senescence and its implications in aging. Genes (Basel) https://doi.org/10.3390/genes8120343 (2017).
Tasdemir, N. et al. BRD4 connects enhancer remodeling to senescence immune surveillance. Cancer Discov. 6, 612–629 (2016).
Franceschi, C. & Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci. 69, S4–S9 (2014).
Stojanovic, S. D., Fiedler, J., Bauersachs, J., Thum, T. & Sedding, D. G. Senescence-induced inflammation: an important player and key therapeutic target in atherosclerosis. Eur. Heart J. 41, 2983–2996 (2020).
Munoz-Espin, D. & Serrano, M. Cellular senescence: from physiology to pathology. Nat. Rev. Mol. Cell Biol. 15, 482–496 (2014).
Xue, W. et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445, 656–660 (2007).
Kang, T. W. et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 479, 547–551 (2011).
Kale, A., Sharma, A., Stolzing, A., Desprez, P. Y. & Campisi, J. Role of immune cells in the removal of deleterious senescent cells. Immun. Ageing 17, 16 (2020).
Kaur, A. et al. sFRP2 in the aged microenvironment drives melanoma metastasis and therapy resistance. Nature 532, 250–254 (2016).
Coppe, J. P., Desprez, P. Y., Krtolica, A. & Campisi, J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu. Rev. Pathol. 5, 99–118 (2010).
Waugh, D. J. & Wilson, C. The interleukin-8 pathway in cancer. Clin. Cancer Res. 14, 6735–6741 (2008).
Ortiz-Montero, P., Londono-Vallejo, A. & Vernot, J. P. Senescence-associated IL-6 and IL-8 cytokines induce a self- and cross-reinforced senescence/inflammatory milieu strengthening tumorigenic capabilities in the MCF-7 breast cancer cell line. Cell Commun. Signal. 15, 17 (2017).
Acosta, J. C. et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 15, 978–990 (2013).
Nelson, G. et al. A senescent cell bystander effect: senescence-induced senescence. Aging Cell 11, 345–349 (2012).
Ovadya, Y. et al. Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat. Commun. 9, 5435 (2018).
Schroth, J., Thiemermann, C. & Henson, S. M. Senescence and the aging immune system as major drivers of chronic kidney disease. Front. Cell Dev. Biol. 8, 564461 (2020).
Prata, L., Ovsyannikova, I. G., Tchkonia, T. & Kirkland, J. L. Senescent cell clearance by the immune system: emerging therapeutic opportunities. Semin. Immunol. 40, 101275 (2018).
Pereira, B. I. et al. Senescent cells evade immune clearance via HLA-E-mediated NK and CD8+ T cell inhibition. Nat. Commun. 10, 2387 (2019).
Teo, Y. V. et al. Notch signaling mediates secondary senescence. Cell Rep. 27, 997–1007 (2019).
Desdin-Mico, G. et al. T cells with dysfunctional mitochondria induce multimorbidity and premature senescence. Science 368, 1371–1376 (2020).
Yousefzadeh, M. J. et al. An aged immune system drives senescence and ageing of solid organs. Nature 594, 100–105 (2021).
Ferreira-Gonzalez, S. et al. Paracrine cellular senescence exacerbates biliary injury and impairs regeneration. Nat. Commun. 9, 1020 (2018).
Grosse, L. et al. Defined p16High senescent cell types are indispensable for mouse healthspan. Cell Metab. 32, 87–99 (2020).
Tchkonia, T., Zhu, Y., van Deursen, J., Campisi, J. & Kirkland, J. L. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J. Clin. Invest. 123, 966–972 (2013).
Xu, M., Tchkonia, T. & Kirkland, J. L. Perspective: targeting the JAK/STAT pathway to fight age-related dysfunction. Pharmacol. Res. 111, 152–154 (2016).
Zhu, Y. et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 15, 428–435 (2016).
Yosef, R. et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun. 7, 11190 (2016).
Tchkonia, T., Palmer, A. K. & Kirkland, J. L. New horizons: novel approaches to enhance healthspan through targeting cellular senescence and related aging mechanisms. J. Clin. Endocrinol. Metab. 106, e1481–e1487 (2021).
Robbins, P. D. et al. Senolytic drugs: reducing senescent cell viability to extend health span. Annu. Rev. Pharmacol. Toxicol. 61, 779–803 (2021).
Kirkland, J. L. & Tchkonia, T. Senolytic drugs: from discovery to translation. J. Intern. Med. 288, 518–536 (2020).
Hernandez-Segura, A., Brandenburg, S. & Demaria, M. Induction and validation of cellular senescence in primary human cells. J. Vis. Exp. https://doi.org/10.3791/57782 (2018).
Amor, C. et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature 583, 127–132 (2020).
Yoshida, S. et al. The CD153 vaccine is a senotherapeutic option for preventing the accumulation of senescent T cells in mice. Nat. Commun. 11, 2482 (2020).
Sallin, M. A. et al. Host resistance to pulmonary Mycobacterium tuberculosis infection requires CD153 expression. Nat. Microbiol. 3, 1198–1205 (2018).
Gustafson, C. E., Kim, C., Weyand, C. M. & Goronzy, J. J. Influence of immune aging on vaccine responses. J. Allergy Clin. Immunol. 145, 1309–1321 (2020).
Yousefzadeh, M. J. et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 36, 18–28 (2018).
Zhu, Y. et al. New agents that target senescent cells: the flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging (Albany NY) 9, 955–963 (2017).
Fan, Y., Cheng, J., Zeng, H. & Shao, L. Senescent cell depletion through targeting BCL-family proteins and mitochondria. Front. Physiol. 11, 593630 (2020).
de Vos, S. et al. Safety and efficacy of navitoclax, a BCL-2 and BCL-XL inhibitor, in patients with relapsed or refractory lymphoid malignancies: results from a phase 2a study. Leuk. Lymphoma 62, 810–818 (2021).
Roberts, A. W. et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 374, 311–322 (2016).
Wilson, W. H. et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 11, 1149–1159 (2010).
He, Y. et al. Inhibition of USP7 activity selectively eliminates senescent cells in part via restoration of p53 activity. Aging Cell 19, e13117 (2020).
Fuhrmann-Stroissnigg, H. et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat. Commun. 8, 422 (2017).
Cherif, H. et al. Curcumin and o-Vanillin exhibit evidence of senolytic activity in human IVD cells in vitro. J Clin. Med. https://doi.org/10.3390/jcm8040433 (2019).
Cai, Y. et al. Elimination of senescent cells by beta-galactosidase-targeted prodrug attenuates inflammation and restores physical function in aged mice. Cell Res. 30, 574–589 (2020).
Munoz-Espin, D. et al. A versatile drug delivery system targeting senescent cells. EMBO Mol. Med. https://doi.org/10.15252/emmm.201809355 (2018).
Hall, B. M. et al. Aging of mice is associated with p16(Ink4a)- and β-galactosidase-positive macrophage accumulation that can be induced in young mice by senescent cells. Aging (Albany NY) 8, 1294–1315 (2016).
Tavassoly, I. et al. Dynamic modeling of the interaction between autophagy and apoptosis in mammalian cells. CPT Pharmacometrics Syst. Pharmacol. 4, 263–272 (2015).
Tai, H. et al. Autophagy impairment with lysosomal and mitochondrial dysfunction is an important characteristic of oxidative stress-induced senescence. Autophagy 13, 99–113 (2017).
Nogueira-Recalde, U. et al. Fibrates as drugs with senolytic and autophagic activity for osteoarthritis therapy. EBioMedicine 45, 588–605 (2019).
Bharath, L. P. et al. Metformin enhances autophagy and normalizes mitochondrial function to alleviate aging-associated inflammation. Cell Metab. 32, 44–55 (2020).
Kucheryavenko, O., Nelson, G., von Zglinicki, T., Korolchuk, V. I. & Carroll, B. The mTORC1-autophagy pathway is a target for senescent cell elimination. Biogerontology 20, 331–335 (2019).
Wakita, M. et al. A BET family protein degrader provokes senolysis by targeting NHEJ and autophagy in senescent cells. Nat. Commun. 11, 1935 (2020).
Kang, H. T. et al. Chemical screening identifies ATM as a target for alleviating senescence. Nat. Chem. Biol. 13, 616–623 (2017).
Triana-Martinez, F. et al. Identification and characterization of cardiac glycosides as senolytic compounds. Nat. Commun. 10, 4731 (2019).
Guerrero, A. et al. Cardiac glycosides are broad-spectrum senolytics. Nat. Metab. 1, 1074–1088 (2019).
Johmura, Y. et al. Senolysis by glutaminolysis inhibition ameliorates various age-associated disorders. Science 371, 265–270 (2021).
Justice, J. N. et al. Senolytics in idiopathic pulmonary fibrosis: results from a first-in-human, open-label, pilot study. EBioMedicine 40, 554–563 (2019).
Hickson, L. J. et al. Corrigendum to ‘Senolytics decrease senescent cells in humans: preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease’ EBioMedicine 47 (2019) 446–456. EBioMedicine 52, 102595 (2020).
Dolgin, E. Send in the senolytics. Nat. Biotechnol. 38, 1371–1377 (2020).
Jeon, O. H. et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 23, 775–781 (2017).
Saltzman, B. M. et al. The therapeutic effect of intra-articular normal saline injections for knee osteoarthritis: a meta-analysis of evidence level 1 studies. Am. J. Sports Med. 45, 2647–2653 (2017).
Omori, S. et al. Generation of a p16 reporter mouse and its use to characterize and target p16high cells in vivo. Cell Metab. 32, 814–828 e816 (2020).
Liu, J. Y. et al. Cells exhibiting strong p16INK4a promoter activation in vivo display features of senescence. Proc. Natl Acad. Sci. USA 116, 2603–2611 (2019).
Burd, C. E. et al. Monitoring tumorigenesis and senescence in vivo with a p16INK4a-luciferase model. Cell 152, 340–351 (2013).
Wang, B. et al. An inducible p21-Cre mouse model to monitor and manipulate p21-highly-expressing senescent cells in vivo. Nat. Aging https://doi.org/10.1038/s43587-021-00107-6 (2021).
Zhou, Y. et al. Senolytics improve bone forming potential of bone marrow mesenchymal stem cells from aged mice. NPJ Regen. Med. 6, 34 (2021).
Acknowledgements
We thank G. Hargis for help with drafting figure images and I. Al-Naggar for feedback during the review process. This Review was supported in part by a Glenn Foundation for Medical Research and AFAR Grant for Junior Faculty (M.X.), the Esperance Fellowship in Personalized Nutrition (N.S.G.), The Kenneth and Paula Munson Family Fund for Student Support in Health Sciences Fellowship (N.S.G.), Robert and Arlene Kogod (J.L.K.), the Connor Group (J.L.K.), Robert J. and Theresa W. Ryan (J.L.K.), the Noaber Foundation (J.L.K.), Travelers Chair in Geriatrics and Gerontology (G.A.K.), and NIH grants R37AG013925 (J.L.K.), R01AG072301 (J.L.K), P01AG062413 (J.L.K.), R33AG061456 (Translational Geroscience Network; J.L.K. and G.A.K.), R21AG063528 (M.X. and G.A.K.), R03AG072374 (M.X.), R01AG066679 (M.X.) and R01AG068860 (M.X.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Patents on senolytic drugs and their uses are held by Mayo Clinic. This research has been reviewed by the Mayo Clinic Conflict of Interest Review Board and was conducted in compliance with Mayo Clinic Conflict of Interest policies.
Additional information
Peer review information Nature Aging thanks Manuel Collado, Ana O’Loghlen and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Gasek, N.S., Kuchel, G.A., Kirkland, J.L. et al. Strategies for targeting senescent cells in human disease. Nat Aging 1, 870–879 (2021). https://doi.org/10.1038/s43587-021-00121-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s43587-021-00121-8
This article is cited by
-
Emerging role of senescent microglia in brain aging-related neurodegenerative diseases
Translational Neurodegeneration (2024)
-
Lessons from inducible pluripotent stem cell models on neuronal senescence in aging and neurodegeneration
Nature Aging (2024)
-
Unleashing CAR T cells to delay metabolic aging
Nature Aging (2024)
-
Kdm6a-CNN1 axis orchestrates epigenetic control of trauma-induced spinal cord microvascular endothelial cell senescence to balance neuroinflammation for improved neurological repair
Bone Research (2024)
-
Targeting aging and age-related diseases with vaccines
Nature Aging (2024)
