
Abstract
Protein glycosylation involves the co-translational or post-translational addition of glycans to proteins and is a crucial protein modification in health and disease. The aim of glycoproteomics is to understand how glycosylation shapes biological processes by understanding peptide sequences, glycan structures and sites of modification in a system-wide context. Over the past two decades, mass spectrometry (MS) has emerged as the primary technique for studying glycoproteins, with intact glycopeptide analysis — the study of glycopeptides decorated with their native glycan structures — now a preferred approach across the community. In this Primer, we discuss glycoproteomic methods for studying glycosylation classes, including best practices and critical considerations. We summarize how glycoproteomics is used to understand glycosylation at a systems level, with a specific focus on N-linked and O-linked glycosylation (both mucin-type and O-GlcNAcylation). We cover topics that include sample selection; techniques for protein isolation, proteolytic digestion, glycopeptide enrichment and MS fragmentation; bioinformatic platforms and applications of glycoproteomics. Finally, we give a perspective on where the field is heading. Overall, this Primer outlines the current technologies, persistent challenges and recent advances in the exciting field of glycoproteomics.
Similar content being viewed by others
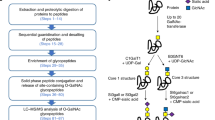
Introduction
Protein glycosylation refers to the covalent attachment of carbohydrates to polypeptides and represents a class of prevalent and structurally diverse co-translational and post-translational modifications (PTMs) that impact a huge number of biological processes1,2,3,4,5,6. Carbohydrate modifications include single monosaccharides and complex carbohydrate chains, both referred to as glycans. Protein glycosylation is a non-templated process and is mediated by enzymes known as glycosyltransferases, responsible for the initiation or elongation of glycans, and oligosaccharyltransferases, responsible for the addition of whole carbohydrate chains. In cells, the complex interplay between glycosyltransferases or oligosaccharyltransferases, carbohydrate transporters and glycosidases — the enzymes that remove these carbohydrates — fine-tunes the glycan structures observed on individual proteins and regulates glycoprotein function, with effects on biological processes that include cellular development7, cell–cell communication8, host–microorganism interactions9,10 and immunity5,11,12. For example, the recruitment of leukocytes to sites of inflammation is precisely controlled by specific glycan structures that mediate interactions with cell-surface lectins to enable selective and site-specific leukocyte homing5,7,11,12. Dysregulation of glycosylation is associated with numerous diseases, including cancer13,14,15,16, infection and inflammation17,18,19,20,21,22, schizophrenia23 and a wide range of congenital and neurological disorders24,25,26. Unravelling the role of glycosylation under both physiological and pathophysiological conditions is a long-standing goal of glycobiology and has driven the rapid development of methods to track glycosylation for diagnostic and therapeutic purposes27,28.
Glycosylation is a universal protein modification across all domains of life with structurally distinct subclasses and glycan types now recognized29,30,31,32,33,34 (Fig. 1a,b). Our knowledge of mammalian asparagine-linked (N-linked) and serine/threonine-linked (O-linked) glycans is the most developed, and these modifications are therefore the focus of this Primer. Characterizing the glycoproteome involves the identification of glycoproteins as well as definition of the macroheterogeneity (structural diversity owing to the presence or absence of glycans at specific glycosylation sites) and microheterogeneity (structural diversity of glycosylation patterns at individual glycosylation sites)35 within these proteins. Microheterogeneity can arise through differences in the number and type of individual monosaccharide residues within the glycan, the structural arrangements and branching patterns of these monosaccharides or the configuration of anomeric linkages (see Box 1 for a guide to the symbol nomenclature for glycans). Ultimately, identifying glycosylation sites and discrete glycan structures is crucial for understanding the roles of glycan-dependent functions in biological processes.

a | A range of glycosylation types exist, with most eukaryotic cells possessing multiple pathways for protein glycosylation. Glycosylation involves the installation of glycans on proteins, with N-linked pathways targeting the nitrogen of asparagine residues, O-linked pathways targeting the oxygen atoms of serine/threonine residues and C-linked pathways targeting the second carbon of tryptophan residues. Many of these glycosylation events are observed on proteins known to be secreted or displayed extracellularly, as denoted here, owing to the role of glycosylation in mediating extracellular protein stability and membrane protein recognition. Intracellularly, O-GlcNAcylation has a crucial role in cellular signalling events. b | A range of common glycan classes is observed across mammalian N-linked and mucin-type O-linked glycosylation. N-linked glycans include paucimannose, oligomannose, and complex and hybrid structures. Paucimannose carries one to three mannose (Man) residues on a chitobiose core with variable core fucosylation. Oligomannose glycans contain terminal branches composed only of mannose sugars. Complex and hybrid glycans may contain galactose (Gal), N-acetylglucosamine (GlcNAc), N-acetylgalactosamine (GalNAc), fucose (Fuc), N-acetylneuraminic acid (NeuAc) and N-glycolylneuraminic acid (NeuGc) residues in their antennae, with hybrid glycans also containing unsubstituted terminal mannose residues. Eight core structures have been described for mucin-type O-linked glycosylation, which differ in their composition and linkage position of branches to a protein-linked GalNAc. Non-canonical glycans introduced using metabolic oligosaccharide engineering approaches are also possible; for non-canonical glycans, the presence of monosaccharides bearing chemical handles such as alkyne or azide (N3) groups allow glycan-specific labelling and/or enrichment. GlcA, glucuronic acid; Xyl, xylose.
Glycoproteomics refers to the systems-level study of protein-linked glycans and is a rapidly evolving analytical field that aims to profile glycosylation events observed within biological samples36,37. The characterization of intact glycopeptides is an attractive analytical strategy as only intact glycopeptides can provide direct evidence of the site-specific glycosylation of proteins. Bottom-up glycoproteomics using liquid chromatography–tandem mass spectrometry (LC–MS/MS)-based profiling of intact glycopeptides allows for cell-wide, tissue-wide and organism-wide mapping of glycosylation events and the ability to address their functional roles in biological processes38. This is in contrast to commonly used techniques that involve the study of detached glycans — a field known as glycomics39 — or formerly N-linked glycosylated peptides (N-glycosylation site mapping40).
LC–MS/MS-driven glycoproteomic approaches have been refined considerably over the past decade and these strategies are increasingly being used for quantitative mapping of glycosylation sites within complex mixtures (as previously reviewed36,38,41,42,43,44,45,46,47,48,49,50,51). Technological and computational advances now enable the characterization of thousands of intact N-glycopeptides and O-glycopeptides within a given glycoproteomics experiment52,53,54,55,56,57,58,59,60. Although analytical challenges still exist61,62,63, this Primer aims to illustrate the technologies, tools and approaches available to address pending questions in glycobiology. By presenting developments across the entire glycoproteomics workflow, this Primer is designed to summarize the field as it currently stands. We cover various biological models, chemical glycobiology approaches, glycopeptide enrichment techniques, quantification strategies, glycopeptide separation and ionization, tandem mass spectral analysis, computational tools for glycopeptide identification and options for data storage and dissemination. We hope this Primer serves as a springboard for anyone entering the field of glycoproteomics.

Experimentation
A multitude of experimental pipelines have been developed for glycoproteomic studies that share several key steps. These steps include sample selection, sample preparation, including protein clean-up approaches, the enzymatic digestion of samples to enable access to desired glycopeptides, separation of glycopeptides from non-glycosylated peptides and analysis of glycopeptides using MS strategies. As we discuss these steps below it should be noted that these steps provide a modular framework and, depending on the glycoproteome studied, can be omitted or altered to enhance the identification of the glycopeptides of interest. Although a range of approaches and preparation pipelines exist to study glycoproteomes, we note that the optimal approach is likely to be different for each biological question, and trials of multiple preparation approaches may be needed to achieve the desired outcome.
Choice of sample
State-of-the-art glycoproteomic workflows are capable of handling complex samples derived from cultured cells, tissues, organs and even whole organisms64,65,66,67,68. The choice of sample will affect the degree of sample processing needed (Table 1). For a given sample, the depth of analysis required is dependent on the total number of proteoforms present and the relative abundance and dynamic range of glycoproteins within the sample. For samples of low complexity, glycosylation analysis can be accomplished with low microgram levels of material, although milligram amounts may be needed for complex samples in which the glycoproteins of interest are present in low concentrations. In general, samples of low complexity with a high glycoprotein abundance will allow for better characterization of glycosites and glycoforms, which underpins the rationale for separating or enriching glycoproteins or glycopeptides before analysis (see below)69,70,71,72.
Biological relevance is important to consider if analysing recombinant glycoproteins from different sources. The observed glycosylation sites and glycan structures of proteins heterologously expressed under in vitro conditions, such as in genetically modified immortalized cell lines, may differ from in vivo sources as the repertoire of expressed glycosyltransferases and glycosidases can vary between cell types32. This is evident for viral envelope glycoproteins such as the HIV-1 envelope protein (Env) and SARS-CoV-2 spike glycoprotein, where higher degrees of N-glycan processing are found on native virions than ectopic expression of individual viral proteins in cell lines73. Furthermore, there can be notable differences in glycosite occupancy and glycan structure between native oligomeric proteins and individually expressed subunits, likely influenced by differences in the accessibility of the subunits and the protein quaternary structure to glycosyltransferases69,70,74,75. Thus, care should be taken to ensure that the models used reflect the biological question being explored as closely as possible.
The redundant and overlapping specificities of glycosyltransferases have profound impacts on glycosylation patterns, as compensation and competition for substrates can make the observed relationships between glycosyltransferases and glycosylation events highly context dependent even across similar cell types. This is best illustrated for O-linked, mucin-type glycosylation, which is governed by the expression of several members of a large family of GalNAc-transferase (GalNAc-T) isoforms6. A diverse array of biological specimens have been probed to study the breadth of the O-glycoproteome53,66,67,68,76,77,78. The competition for substrates between GalNAc-T isoforms is complex and largely unclear, and genetically engineered cell lines have been used to dissect substrates of specific GalNAc-T isoforms79,80. Further, isogenic cell lines and transgenic animal models generated using gene editing have identified GalNAc-T isoform-specific substrates in the context of both simplified and natural glycan structures79,81,82,83. These findings highlight the benefits of genetic approaches for understanding glycosylation site specificity in situations in which complex interplays exist. Considering this known complexity associated with glycosylation substrates for many glycosylation systems, it is advisable to include several biological replicates representing different clonal lineages of genetically engineered cell lines and only consider consistent changes relevant83,84.
Sample preparation
Protein isolation and buffer considerations
Optimal protein isolation is key for efficient downstream sample processing in all proteomic experiments. Protein extraction from tissues can require pre-treatment with enzymes or ethylenediaminetetraacetic acid (EDTA) to release cells from the extracellular matrix before cell lysis. Once isolated, cells can be lysed with cryogenic homogenization, mechanical disruption using sonication or mechanical grinding in buffers that contain strong detergents such as sodium dodecyl sulfate (SDS) or chaotropic agents85,86,87,88. Complex tissue-derived and cell-derived samples will rarely be solubilized completely and often require clearing of the lysates by centrifugation to remove insoluble material. Homogenization may also be necessary for viscous biological secretions such as sputum or intestinal mucus89,90. It should be noted that several commonly used cationic, anionic or zwitterionic detergents can interfere with proteolytic digestion and may cause LC–MS analyte signal suppression without subsequent clean-up (see below)91,92. MS-compatible detergents such as RapiGest76,93,94, N-dodecyl β-d-maltoside95 or ProteaseMAX96 have been used for glycoproteomic studies to solubilize membrane proteins and can be combined with orthogonal isolation methods such as mechanical disruption to enhance protein isolation77,97. Notably, these MS-compatible detergents can be less effective solubilization agents than strong detergents such as SDS98. The isolation of membrane-bound glycoproteins requires vigorous disruption of the cell membrane followed by a solubilization step that uses detergents or chaotropic agents to prevent the precipitation of hydrophobic proteins99; for soluble secreted glycoproteins, the most important consideration when preparing the sample is to avoid contamination from exogenous protein sources commonly used to maintain cell lines, such as fetal bovine serum, which can be achieved by briefly culturing cells in serum-free medium100.
For many glycoproteomic studies, it may be essential to ensure complete linearization of glycoproteins during solubilization by removing disulfide linkages with the aid of reduction agents such as dithiothreitol (DTT) or tris(2-carboxyethyl)phosphine (TCEP). Ensuring protein linearization can improve the ability of detergents to coat hydrophobic regions within glycoproteins; however, this process also results in the generation of reduced cysteine residues, which are extremely reactive and readily undergo oxidation as well as other chemical transformations. Alkylation of reduced cysteines can ‘cap’ these reactive amino acids, preventing the formation of undesirable cysteine products and the re-formation of disulfide linkages during sample preparation. Iodoacetamide is commonly used to alkylate cysteine residues during glycoproteomic sample preparation. Although alkylation is advantageous for improving the detection of cysteine-containing peptides, it has been noted that the underalkylation or the unintended alkylation of residues such as methionine (overalkylation) can cause the misassignment of glycan compositions, as these events unexpectedly change the glycopeptide mass to match isobaric alternative glycan compositions, leading to incorrect glycopeptide assignment61. Both glycoproteomic61 and proteomic101 studies have highlighted that underalkylation and overalkylation are commonplace, and care should be taken to ensure that alkylation reagent concentrations and incubation times are optimized for the given sample.
Glycoproteome clean-up approaches
To facilitate the analysis of chemically solubilized samples, recent advancements in sample preparation offer attractive solutions to removing interfering chemical agents such as salt and detergents before subsequent MS analysis. Three such approaches are filter-aided sample preparation (FASP)102, suspension traps (S-traps)103,104 and methods based on protein aggregation capture (PAC)105,106,107,108,109 (Fig. 2). These methods involve binding proteins to solid-phase supports such as filters (FASP), quartz mesh (S-traps) or magnetic particles (PAC) and washing with chaotropic agents or organic solvents to remove contaminants; digestion of the bound proteins then releases peptides for subsequent analysis. FASP-based sample preparation is well established and has been implemented in numerous N-glycoproteomic studies across species and tissues64,110, whereas S-traps and PAC-based approaches such as single-pot, solid-phase-enhanced sample preparation (SP3)111 are a more recent addition to the glycoproteomics toolkit (although they have been implemented in several glycoproteomic studies)112,113,114. These approaches can be used for sample amounts as low as a few micrograms to several milligrams of protein, and they result in high peptide recovery rates102,103,104,111. It was recently demonstrated that PAC enables the removal of chemical or affinity tag agents typically used in click-based labelling105,106, making PAC particularly appealing for bioorthogonal glycoproteomic sample preparation.

Glycoproteomic sample preparation can be summarized into six key steps. a | Proteins for glycoproteomic analysis are extracted and solubilized from samples of interest such as from cell culture models using a cell disruptor to lyse the cells. b | Protein mixtures are processed to remove potential interfering reagents for downstream processing with filter-aided sample preparation (FASP), quartz mesh (S-trap) and protein aggregation capture (PAC)-based approaches commonly used. c | The resulting protein preparations are then digested with proteases and/or glycoproteases to generate mixtures that contain the glycopeptides of interest for downstream analysis. Digestion of FASP, S-trap or PAC prepared samples allows the release of peptides from the captured proteins enabling their collection for downstream liquid chromatography–mass spectrometry (LC–MS) analysis. At this stage, glycosidases can also be used to remove specific glycans of interest or modify glycans to enhance their downstream detection by reducing microheterogeneity. d | The resulting peptide mixtures containing the glycopeptides of interest can be concentrated and purified, allowing the removal of non-digested proteins, enzymes or buffer components that may interfere with chemical labelling or enrichment approaches. Several solid-phase clean-up media can be used to achieve this, including C18, hydrophilic–lipophilic balance (HLB) or styrenedivinylbenzene–reverse phase sulfonate (SDB–RPS) resins, which can be implemented in solid-phase extraction (SPE) cartridge, plate or microcolumn (Zip/STAGE tips) formats. e | Further peptide-based chemical derivatization can be undertaken to enable enrichment, quantification or to enhance the detection of glycopeptides during downstream LC–MS analysis. For example, the incorporation of positively charged imidazolium groups within biotin-based enrichment handles can be used to improve electron-driven dissociation (ExD)-based fragmentation. f | Glycopeptides of interest can be enriched using affinity approaches before LC–MS analysis, such as streptavidin enrichment of biotin-labelled metabolic ogligosaccharide engineering (MOE) samples, lectin weak affinity chromatography (LWAC), which exploits the binding of lectins to specific sugars, or hydrophilic interaction liquid chromatography (HILIC), which retains glycopeptides based on hydrophilic interactions.
Proteome digestion approaches
After clean-up, glycoproteins can be digested using proteases to produce individual peptides and glycopeptides (Fig. 2). The conversion of proteins into (glyco)peptides offers a range of analytical advantages in both downstream separation and mass spectral analysis. Reducing the chemical heterogeneity of a proteome to a mixture of soluble peptides enables separation with much higher resolution than intact proteins. Furthermore, smaller peptides fragment more efficiently and produce simpler spectra, aiding the characterization of modification sites. The workhorse protease for glycoproteomics is trypsin, which cleaves at the C terminus of arginine or lysine residues with high specificity, efficiency and robustness. This generates peptides that can be protonated at the amine-containing N terminus and the arginine/lysine residue at the C terminus, resulting in rich MS/MS spectra when analysed in positive polarity mode. Although trypsin is the protease of choice for most N-glycoproteomic and O-glycoproteomic analyses, O-glycosites are commonly found in dense clusters notoriously resistant to tryptic cleavage owing to a lack of arginine/lysine residues96, which limits the applicability of trypsin to these densely O-glycosylated domains. To address this issue, many groups have employed digestion with several alternative proteases that possess different cleavage specificities to increase proteome coverage, such as chymotrypsin to cleave C-terminally to phenylalanine, tryptophan and tyrosine; GluC, which cleaves C-terminally to glutamic acid and to a lesser extent aspartic acid, or AspN, which cleaves N-terminally to aspartic acid and to some extent glutamic acid72,115,116,117.
Non-specific proteases such as Pronase and Proteinase K have also been used to analyse a range of glycosylated proteins. Pronase is a commercially available mixture of proteases isolated from Streptomyces griseus that exhibits both exoprotease and endoprotease activities and yields a crude mixture of heterogeneous peptide fragments118. Pronase is useful for the glycoproteomic analysis of samples of modest complexity119; however, the peptide heterogeneity generated by Pronase digestion is a major issue for quantitative site-specific glycan profiling. Similar to Pronase, Proteinase K is an endoprotease that cleaves at the C termini of aliphatic and aromatic residues and is often used in conjunction with trypsin digestion for glycosylation site localization of simple mixtures120. The drawback of both non-specific digestion techniques is that the resultant data must be searched against all theoretical peptides, producing an extremely large search space that increases search time and false discovery rates (FDRs; discussed below)121. Further, the propensity of these proteases to generate relatively short glycopeptides limits their usefulness for complex samples, as mapping the identified glycopeptides to specific proteins can be difficult. Thus, the use of non-specific proteases is typically restricted to single-protein mixtures, where this approach is most appropriately used to characterize regions such as mucin domains that cannot be accessed by other enzymes122. It should also be noted that despite these challenges, the high levels of peptide heterogeneity observed with these enzymes can be advantageous for applications such as the localization of glycosylation events to specific amino acids119,120,122.
Glycoproteome-centric proteases (O-glycoproteases)
Glycoproteases are increasingly being used in O-linked glycoproteomic studies123. O-glycoproteases have modest peptide sequence specificities, cleaving the peptide backbone based on the presence of various O-linked glycans and allowing the digestion of glycosylated regions resistant to other proteases. OgpA, derived from Akkermansia muciniphila and marketed and sold as OpeRATOR, was the first commercial O-glycoprotease. This enzyme cleaves at the N terminus of serine or threonine residues that bear truncated glycans such as GalNAc or GalNAc-Gal, also known as core 1 O-glycans (Fig. 1b). OgpA has been used for the digestion of isolated O-glycoproteins, cell lysates and tissues56,124. Its main drawback is that it is unable to cleave glycopeptides decorated with sialic-acid-containing O-glycans; thus, samples must be sialidase-treated before proteolytic digestion. Additionally, OgpA can be inefficient in regions that are densely glycosylated, requiring downstream electron-based fragmentation for confident O-glycosite localization63.
Several glycoproteases other than OgpA have been introduced to the field. Secreted protease of C1 esterase inhibitor (StcE), derived from enterohaemorrhagic Escherichia coli, is specific for a serine/threonine*-X-serine/threonine motif, cleaving before the second serine/threonine (the asterisk indicates that the first serine/threonine is invariably glycosylated). StcE improved the analysis of densely O-glycosylated mucin-domain glycoproteins, increasing protein sequence coverage, the number of glycosites identified and the number of localized glycans in proteins studied96. Expanding on this concept exploiting the diversity of bacterial glycoproteases as glycoproteomic tools, the Bertozzi group compiled a glycoprotease toolkit of six additional enzymes: Bacteroides thetaiotaomicron 4244 (BT4244), A. muciniphila 0627 (AM0627), 1514 (AM1514) and 0608 (AM0608), enteroaggregative E. coli protease involved in colonization (Pic), and Streptococcus pneumoniae zinc metalloprotease C (ZmpC), where each has a different cleavage motif125. Similarly, other groups have demonstrated that enzymes such as the coagulation-targeting metalloendopeptidase (CpaA) of Acinetobacter baumannii126 and the immunomodulating metalloprotease (IMPa) from Pseudomonas aeruginosa also cleave glycosylated serine and threonine residues with unique specificities127.
Endoglycosidases and exoglycosidases
Endoglycosidases release oligosaccharides from the protein attachment site or within the glycan chain, whereas exoglycosidases trim monosaccharides from the non-reducing termini of the glycan chain128. The removal of glycans or the reduction of glycan heterogeneity can concentrate the observable signal of glycosylated or previously glycosylated peptides to a limited number of chemical species, which can enhance the detection of glycosylation events. One of the most commonly used endoglycosidases is PNGase F, which cleaves intact N-glycans from proteins and deamidates the previously modified asparagine residue to aspartic acid. Similar enzymes such as Endo F and Endo H cleave within the chitobiose N-glycan core to leave a single GlcNAc on the modified asparagine residues129,130. A universal endo-O-glycosidase has not been characterized, although some glycosidases can remove truncated O-glycan structures, for example, OglyZOR, a commercially available endoglycosidase derived from Streptococcus oralis that hydrolyses truncated core 1 O-glycans. Commercial glycosidases derived from S. pneumoniae and Enterococcus faecalis that release core 1 and (to a limited extent) core 3 O-glycans are also available. Many O-glycosidases have limited activity if the glycans are modified by sialic acid or GlcNAc and thus must be used in conjunction with other glycosidases to remove these modifications44.
Exoglycosidase treatment is commonly used to simplify glycoproteomic analyses. Sialidases are often used to remove sialic acids, reduce microheterogeneity and limit the number of detected glycoforms, which can improve the identification of glycopeptides131. Broad-acting sialidases such as neuraminidase A can remove sialic acid residues α2,3, α2,6 or α2,8 linked to a glycan, whereas some sialidases are specific for a particular linkage; for example, Clostridium perfringens neuraminidase is commonly used to cleave α2,3 linkages78. Other exoglycosidases used in O-glycoproteomics include β1,4-galactosidase from S. pneumoniae, which removes β1,4-linked galactose, and β-N-acetylhexosaminidase — also from S. pneumoniae — which removes terminal non-reducing HexNAc residues from oligosaccharides49. Owing to the innate specificity of these enzymes, exoglycosidases are useful for trimming glycans for targeted characterization of glycan epitopes and simplifying glycoproteomic analysis. However, removing monosaccharides does limit the information that can be gleaned using intact glycoproteomics.
Chemical and biological affinity-based glycopeptide enrichment
In-depth glycoproteomic analysis benefits from selective enrichment of glycopeptides with affinity-based approaches broadly used across the field and are classified as being chemical or biological in nature. Within this section we introduce common protocols for N-glycopeptide and O-glycopeptide enrichment yet highlight that for a detailed discussion of the breadth of glycopeptide enrichment approaches used across the community readers are referred to exhaustive literature on this topic36,41,43,129,132.
Some of the first proteome-scale studies of glycosylation events used chemical enrichment strategies such as the covalent tethering of glycoproteins or glycopeptides to hydrazide-based resins through cis-diols within the carbohydrate chains. These approaches allow the formation of covalent linkages between resins and the glycopeptides or glycoproteins of interest and allow the removal of non-glycosylated peptides or proteins with detergents or chaotropic agents followed by the elution of the enriched glycopeptides by enzymatic or chemical cleavage of the linked glycans133,134,135,136,137,138,139,140,141. The need to release N-glycans of glycopeptides using PNGase F or the acid hydrolysis of hydrazide-linked sialic acids in these methods has led to the development of alternative chemical enrichment approaches that do not require the removal or alteration of glycan structures. For example, several boronic acid-based resins have been developed that allow glycopeptide enrichment using reversible covalent tethering of glycopeptides142. Additionally, many approaches have been developed that exploit charge-based interactions, including the capture of glycopeptides carrying terminal acidic sugars (such as NeuAc) using titanium dioxide143,144,145 and electrostatic repulsion–hydrophilic interaction chromatography (ERLIC)146. Not all glycans are charged, and several approaches that exploit the hydrophilic nature of glycans have also been developed for various classes of glycopeptides, such as hydrophilic interaction liquid chromatography (HILIC)147,148,149,150 (Fig. 2). Chemical enrichment approaches can typically be undertaken without the need for genetic or metabolic manipulation of models with commercial reagents, and these approaches are therefore applicable to a wider range of biological systems.
In contrast to chemical approaches, naturally occurring proteins that recognize carbohydrate epitopes can also be used for glycopeptide enrichment. A widely used class of carbohydrate-recognizing proteins are lectins, which can be used in lectin weak affinity chromatography (LWAC; Fig. 2) set-ups to enable the enrichment of different subtypes of glycopeptide using a diverse array of commercially available lectins — such as wheat germ agglutinin (WGA) and jacalin lectins, which recognize O-GlcNAc and core 1 O-glycans, respectively80,116,151,152,153,154. LWAC approaches involve the use of lectins immobilized to solid supports, such as agarose, which enable the retention of glycopeptides and the removal of non-glycosylated peptides by washing with mild non-denaturing buffers155. WGA-based LWAC is a common O-GlcNAc enrichment technique, although recent work suggests that commercial anti-O-GlcNAc antibody mixtures are more selective and specific for O-GlcNAcylated peptides114,156. An alternative for core 1 O-GalNAc glycoproteomics is peanut agglutinin (PNA) lectin53,66,68. Vicia villosa agglutinin (VVA) is also well suited for the enrichment of glycopeptides that bear a single O-GalNAc (Tn, Fig. 1b); this lectin was implemented into the SimpleCell O-glycoproteomics approach, where cultured cells are genetically engineered to express homogeneous O-GalNAc glycosylation76,77. Both LWAC and antibody-based enrichment allow glycopeptides to be isolated and eluted with competitive free-carbohydrate solutions155 or through denaturation of the affinity protein with acid114. In addition to its use in studying N-linked and O-linked glycosylation, LWAC-based enrichment has also been applied to study O-Man glycosylation. LWAC-based enrichment of O-Man glycopeptides has been achieved using concanavalin A (ConA) lectin, which recognizes O-linked, but not C-linked, α-mannose sugars94,157,158. It is important to note that the broad and poorly defined specificities of most lectins can complicate interpretation of glycopeptide enrichment results and care must be taken when interpreting glycans enriched with a given lectin.
Metabolic engineering of oligosaccharides for glycopeptide enrichment
Metabolic oligosaccharide engineering (MOE; Fig. 2) has emerged as an important strategy to profile N-glycans and O-glycans58,93,159,160. In MOE, monosaccharides are chemically modified with tags and incorporated into proteins with endogenous glycosylation machinery. The tags are stable in the cellular environment, but reactive against bioorthogonal click chemistry strategies, such as copper-mediated azide-alkyne cycloaddition161. The addition of ‘clicked’ functionalized biotin allows tagged glycopeptides to be enriched using streptavidin-conjugated beads before MS analysis129,162. Metabolic incorporation of clickable alkyne- or azide-modified sugars has been demonstrated for mapping N-glycosites93 and O-GalNAc163,164,165 or O-GlcNAc proteomes166,167. One benefit of MOE is that the functionalized glycans can be incorporated into glycan structures without a chain-terminating effect, allowing additional sugars to be added by endogenous glycosyltransferases. However, labelling efficiency in MOE is extremely low, and reagents are of limited specificity as they can be interconverted and incorporated into unintended glycan structures. A bump-and-hole strategy can be used to label cellular glycans with engineered GalNAc-Ts that accept bumped GalNAc donors168,169,170, delineating GalNAc-T specificities. This strategy has been further developed using a metabolic labelling probe (GalNAzMe) for specific labelling of O-glycans171, as well as clickable tags (ITag) that stably increase glycopeptide charge172.
Analysis of glycopeptides
Glycopeptides are typically characterized using LC–MS/MS, whereby glycopeptides eluted from an LC column are ionized by electrospray ionization (ESI) and sequenced using a suite of tandem MS (MS/MS) dissociation methods41,48,49. Parameters for LC and MS/MS stages are key decision points in glycoproteomic experiments and ultimately have consequences for data quality and interpretation. Matrix-assisted laser desorption/ionization (MALDI)–MS is also a popular high-throughput approach for glycopeptide analysis, although the ability to automate ESI and directly couple it to separation technologies allows a greater dynamic range for complex samples and has made ESI-based LC–MS/MS the mainstay of most glycoproteomic methods. ESI-based LC–MS/MS strategies are therefore the focus of this section.
Liquid chromatography-based separation of glycopeptides
Most glycoproteomic methods use low-pH (pH <2) reverse phase liquid chromatography (RP-LC) to separate glycopeptides before MS/MS, with a C18-based stationary phase and flow rates that range from tens to hundreds of nanolitres per minute (nanoflow). RP-LC is a versatile and robust method widely used in proteomics as it offers a combination of high peak capacity and simplicity173. The retention and thus separation of glycopeptides in the RP-LC column is mostly driven by the hydrophobicity of the peptide backbone, although the size, conformation and monosaccharide content of glycans also contribute to retention behaviour174,175,176. Retention times are useful for glycopeptide identification in combination with the accurate precursor mass and tandem MS spectra, especially when ambiguous MS/MS spectra generate several potential glycopeptide candidates. Prediction tools can help incorporate this orthogonal information from RP-LC177,178,179, although adoption of these data into informatic tools is not yet ubiquitous.
There is no universal separation technique that is ideal for all classes of glycoconjugates129, and although RP-LC is the dominant separation modality in LC–MS/MS glycoproteomics, it does have some drawbacks, such as the co-elution of isomeric glycoforms owing to their identical peptide sequences180,181,182. Although the use of elevated column temperatures in RP-LC can allow the separation of isomeric N-glycopeptides and O-glycopeptides183, this does not always provide adequate separation of all isomeric species. Alternatively, HILIC-LC, in which separation is largely influenced by the hydrophilicity imparted by glycan moieties, can be used in online glycopeptide separations and is effective at separating isomeric species that differ only in glycan linkage position and branching184,185,186. Several HILIC-LC resins exist187 and new HILIC resins provide novel separation characteristics that may be beneficial for specific glycopeptide classes181. Another RP-LC alternative uses porous graphitized carbon (PGC) as the stationary phase, which retains polar compounds with MS-compatible solvents188 and is highly advantageous for separating released glycans189. Its use for separating glycopeptides is somewhat complicated as both hydrophobicity and charge contribute to retention using this separation modality190,191,192; furthermore, highly sialylated glycopeptides and glycopeptides derived from commonly used proteases such as trypsin, GluC or chymotrypsin are difficult to elute from the resin, meaning non-specific proteases that generate shorter glycopeptides are typically required193,194,195,196,197. PGC-LC has been shown to separate isomeric N-glycopeptides and O-glycopeptides198, and separation of glycopeptides with α2,3-linked or α2,6-linked sialic acids can be modulated by column temperature199. However, challenges with the elution of large glycopeptides owing to the retention of hydrophobic species have limited the widespread use of PGC-LC in LC–MS/MS glycoproteomics. We compare separation techniques in Table 2. It is worth noting that although the above-mentioned LC-based approaches are traditionally performed using columns, they can also be successfully employed using chip-based fluidic devices180.
Non-liquid chromatography-based separation of glycopeptides
Separation techniques other than LC are increasingly finding applications in the fine structural analysis of glycans and glycopeptides38. Online capillary electrophoresis (CE) is an emerging tool for glycoproteomics that can separate glycopeptide isomers and offer potential improvements in reproducibility and sensitivity200,201,202,203. Electrophoretic mobility in CE is governed by glycopeptide charge-to-size ratios, and, as a result, glycan composition (and especially sialic acid content) can affect migration, providing glycan-based separation of glycoforms of the same peptide backbone204,205,206. Gas-phase separations of glycopeptides following LC or CE can also be used to separate isomeric glycopeptides; these techniques include ion mobility spectrometry (IMS) approaches207,208,209,210 such as travelling-wave IMS211,212,213,214,215, differential/high-field asymmetrical waveform IMS216,217,218,219 and drift-tube IMS220,221,222,223. In addition to allowing isomeric separation, IMS has also been shown to enable separation of glycosylated species from non-modified peptides, providing access to glycopeptides incompatible with chromatographic enrichment224,225.
The benefits of individual separation approaches (which are summarized in Table 2) can be leveraged together. Offline separation is typically used to fractionate complex mixtures of glycopeptides — usually enriched before fractionation — into multiple samples, with each sample then analysed by LC–MS/MS using an orthogonal separation modality. This fractionation approach can markedly increase sensitivity by reducing the complexity of the mixture being analysed in each online LC–MS/MS analysis; conversely, this dramatically decreases throughput as the analysis of a single sample is spread across multiple LC–MS/MS acquisitions. One such prominent ‘2D’ glycoproteomic approach is offline high-pH RP-LC followed by online low-pH RP-LC57,142,226,227,228,229,230, although offline fractionation with HILIC-LC, PGC-LC and CE have been used prior to online low-pH RP-LC44,119,231,232. Other combinations of glycopeptide separation techniques can provide unique advantages of separating on both glycan and peptide components182, such as offline RP-LC coupled with online CE203, offline HILIC-LC coupled with offline PGC-LC followed by MALDI–MS233 and offline RP-LC coupled with online HILIC-LC60. Two-dimensional separations can also be achieved fully online through carrying out two orthogonal separations on an LC system coupled to the mass spectrometer (for example, online RP-PGC-MS/MS)121,122,234,235,236. As these methods often require specialized equipment, they are not as widely used as offline fractionation followed by online orthogonal separation with LC–MS/MS.
Tandem MS fragmentation of glycopeptides
Several acquisition approaches are available on modern MS instruments237,238 and the choice of fragmentation method — also referred to as the dissociation method — needed to generate MS/MS spectra is determined by the key information required for glycopeptide identification239. Each fragmentation strategy generates specific fragment ion types that determine what information can be obtained for glycopeptide characterization35,240,241,242 and also dictates the instrument platforms suitable for a given experiment, appropriate data acquisition strategies and the informatic tools available for post-acquisition analysis.
The most ubiquitous fragmentation strategy is collision-induced dissociation, which can be accomplished using beam-type collision-induced dissociation (beamCID) — referred to as higher-energy collisional dissociation (HCD) on some instrument platforms243 — or resonance activation collision-induced dissociation (resonanceCID), which is commonly undertaken using ion traps. BeamCID and resonanceCID have notable differences in the resulting spectra of glycopeptides as a result of their different mechanisms and timescales of collisional energy deposition244. ResonanceCID spectra are typically dominated by fragments resulting from glycosidic cleavages, denoted as B/Y-type ions as per the nomenclature published by Domon and Costello242, whereas beamCID provides access to both glycosidic and amide peptide bond fragmentation events245 with amide peptide bond fragments given as a-, b- and y-type ions according to the nomenclature published by Biemann240. Further, ions with low mass to charge ratio (m/z) are typically lost during resonanceCID, whereas these ions are detectable using beamCID244.
BeamCID has become the preferred collision-induced dissociation approach for glycoproteomics owing to its ability to access both glycan and peptide fragments and high-m/z and low-m/z ions. Additionally, beamCID spectra enable rapid MS/MS acquisition rates, with modern mass spectrometers capable of acquiring more than 20 scans per second. BeamCID collision energies can be adjusted by modulating direct current offsets applied to collision cell devices within mass spectrometers, making collision energy a user-adjustable parameter when designing methods. Lower relative collision energies favour glycan fragments (typically B/Y-type ions and some cross-ring fragments), and higher relative collision energies favour peptide fragments (typically b-type and y-type ions with and without glycan loss)246,247,248,249,250,251. Oxonium ions — relatively low-mass ions derived from monosaccharide and disaccharide fragmentation — are also a dominant feature of beamCID spectra. For N-glycopeptides more so than for O-glycopeptides, beamCID can generate b/y-type ions that retain the initiating HexNAc moiety, which can aid glycosite localization. The generation of b/y-type ions that retain intact glycan species is rare in beamCID regardless of collision energy, although the presence of these ions is more likely for glycopeptides with low proton mobility252,253; lack of b/y-type ions with intact glycan species complicates spectral interpretation and glycosylation localization where multiple potential glycosites are present in a given glycopeptide, a challenge most often encountered with O-glycopeptides36,42,63,254. An emerging trend is the use of stepped-collision-energy beamCID (SCE-beamCID), in which a single MS/MS spectrum is collected for product ions generated using multiple collision energies for the same glycopeptide precursor54,55,247,248,255,256. SCE-beamCID methods often provide multiple types of informative fragment that can aid identification and structural analyses, although this does not ameliorate the weaknesses of beamCID for O-glycosite localization252.
Alternative methods to collision-induced dissociation include those that use electrons or photons as the means of fragmentation257. Electron-driven dissociation (ExD) methods such as electron capture dissociation (ECD) and electron transfer dissociation (ETD) generate c/z-type ions for peptide backbone sequencing (as defined by the Biemann peptide fragmentation nomenclature)240, with little to no fragmentation of glycan moieties. These methods are therefore complementary to beamCID and particularly useful for site-specific characterization of O-glycopeptides and other glycopeptides with multiple potential sites of modification43,77,258,259,260,261. ExD is also valuable for highly charged species, although the generation of sequence-informative fragment ions decreases at low precursor cation charge densities262. This can be problematic for glycopeptide analysis, in which neutral or negatively charged glycans add mass without a concomitant addition of positive charge. Additionally, glycan size and attachment site can affect ExD dissociation owing to secondary gas-phase structure effects263. Hybrid fragmentation methods that combine ExD with collisions (for example, electron transfer/higher-energy collision dissociation, or EThcD) or photons (activated-ion ETD) can address these issues57,241,264,265. Beyond improving fragment ion generation from ExD itself, these hybrid methods also generate fragment ion types from each dissociation mode — for example, in the EThcD regime, c/z-type peptide fragment ions are generated from ETD, and b/y-type peptide fragment ions and B/Y-type glycan fragment ions are generated from beamCID59,241,264,266. Photon-based dissociation methods, particularly ultraviolet photodissociation (UVPD), have also shown promise for generating information-rich spectra with multiple fragment ion types for glycopeptides267,268,269,270, but have yet to be explored for large-scale glycoproteomics.
Although ExD and related hybrid methods can generate high-quality spectra for both N-glycopeptides and O-glycopeptides, these methods often have reaction times of tens to hundreds of milliseconds per spectrum262. BeamCID, by comparison, provides near instantaneous fragmentation. BeamCID or SCE-beamCID methods are therefore more suited for large-scale N-glycopeptide analyses, where b/y-type ions — some of which retain an initiating HexNAc — and B/Y-type ions are mostly sufficient for identification271. Conversely, ExD-centric methods are favourable for O-glycopeptide characterization despite high time costs, as c/z-type ions that retain intact glycan modifications are often necessary for O-glycosite localization59,63,252,258,259,266,272. Experiments that require ExD often combine beamCID and ExD in a product-dependent fashion273,274,275. In product-dependent acquisition schemes, more expedient beamCID methods are used to sequentially fragment precursor ions to look for potential glycopeptides. Once a specific product ion is observed, for example, abundant oxonium ions from a given precursor, the instrument then triggers an ExD spectrum for that same ion, creating complementary pairs of beamCID and ExD spectra for the same precursor ions and relegating ExD spectral acquisition to only those ions that are likely to be glycopeptides.
Glycopeptide data acquisition approaches
Glycoproteomic methods rely heavily on data-dependent acquisition (DDA)38: here, the first mass spectrometer (MS1) scan measures intact glycopeptide ions across a wide m/z range (for example, m/z 400–1,800) as they elute from the LC column and are ionized by ESI. Ions are then isolated using ~1–3 atomic mass unit (amu) windows, fragmented using one of the dissociation strategies discussed above, and the subsequent fragment ions are measured in an MS/MS spectrum with the underlying assumption that fragment ions are largely derived from a single precursor ion. DDA typically prioritizes ions by abundance and sequentially selects analytes for MS/MS analysis, starting with the most abundant and/or desired charge states.
As an alternative to DDA, data-independent acquisition (DIA) isolates large overlapping windows of ions that are designed to cover a user-defined mass range276,277. Each window of ions may contain multiple peptide and glycopeptide species that co-isolate and are thus co-fragmented, and as a result MS/MS spectra contain fragments from multiple precursor ions50. DIA methods iterate over the same windows in a repeating fashion with a defined duty cycle regardless of the signal in MS1 scans, which can aid in sampling of low-abundance ions and improve reproducibility across multiple acquisitions. The complex MS/MS spectra resulting from DIA are challenging to interpret, especially for inherently complex analytes like glycopeptides277. A particular challenge that remains unresolved is the fact that related glycopeptide forms tend to generate near-indistinguishable fragment patterns, making it difficult to identify which precursor structures fragments arise from if captured in the same window. Several DIA methods for glycoproteomics have emerged in recent years278,279,280,281,282,283,284,285,286, and the momentum of DIA in traditional proteomics will likely propel a growth in DIA for glycoproteomics in the future if the above challenge can be overcome50. DIA could be especially beneficial for structure-focused glycoproteomics, as partially resolved, co-eluting glycoforms can be distinguished based on unique chromatogram profiles of fragment ions, enabling quantification of isobaric glycoforms38.
In DDA, the ability to combine several dissociation methods or acquisition styles (for example, product-dependent methods) allows the use of dynamic acquisition schemes that can leverage the strengths of multiple dissociation approaches252. Conversely, DIA requires rapid MS/MS acquisition to enable iterative sampling of all m/z windows across the mass range, which limits the range of dissociation methods that can be implemented efficiently and the ability to dynamically switch between dissociation methods. This limits DIA largely to beamCID-based strategies as ExD spectra simply require too much time to acquire, meaning most glycoproteomic methods that employ DIA to date have focused on simple mixtures of N-glycopeptides278,279,280,281,282,283,284,285. Although O-glycoproteomic studies using DIA have been described, they currently rely on additional DDA-based ExD methods for O-glycosite localization286. Instrumentation that reduces acquisition times for ExD spectra could have the potential to enable ExD-based DIA methods for large-scale glycoproteomics287,288.
Quantification approaches and multiplexing
Several strategies exist for the relative quantification of glycosylation across different samples including those targeted at live cells, proteins or peptides. These methods vary in their multiplexing capacity, quantification accuracy and time and cost effectiveness.
The most common type of quantification is label-free quantification (LFQ). Here, signal intensity or spectral counts are considered to determine relative abundance and each LC–MS analysis corresponds to a single sample, resulting in no sample multiplexing. LFQ analysis has been used to study a range of glycoproteomes including O-GalNAc286 and N-linked glycosylation events289. Although extremely accessible and cost effective, LFQ methods can be less accurate than other methods290.
Stable isotope labelling by amino acids in culture (SILAC) is a highly accurate yet costly method to identify and quantify relative differential changes in complex protein samples291. In this technique, cells are grown in the presence of ‘heavy’ 13C-labelled or 15N-labelled amino acid isotopologues to allow their incorporation into proteins, which leads to an observed mass shift in the MS1 spectrum of labelled peptides. By mixing labelled and unlabelled samples, the relative abundance of peptides or glycopeptides can be determined by comparing the ratio of the light and heavy forms at the MS1 level52,291,292. SILAC typically enables the multiplexing of up to three samples and has been used for N-glycoproteomic studies to understand insulin resistance within adipocytes52, track N-glycan processing and monitor temporal and stress-induced changes in O-GlcNAcylation events156,293. Other stable isotope-based labelling strategies for quantification at the MS1 level include dimethyl294,295 or diethyl296 labelling of peptides, which offers an inexpensive alternative for large-scale experiments and multiplexing of up to three samples296,297. These approaches have been applied for differential glycoproteomic analyses of O-GalNAc and O-Man glycoproteomes, allowing the study of the substrate specificities of GalNAc-Ts79,81 and the mannosyltransferases POMT1 and POMT2 (ref.157) and TMTC1–TMTC4 (ref.158).
A further strategy to enhance multiplexing is the use of isobaric labels that contain different stable isotopes298,299,300 such as isobaric tags for relative and absolute quantification (iTRAQ)301 and tandem mass tags (TMT)298. Upon fragmentation, reporter ions of various masses are generated and their intensities are used for quantification at the MS/MS or MS/MS/MS (MS3) level302,303 with multiplexed analyses of up to 18 samples possible304. An additional advantage of isobaric labelling for glycoproteomics is a notable increase in the observed charge states of glycopeptides, which enhances electron-driven fragmentation305. Despite the advantages, the high price of isobaric labels and the ability to label only submilligram quantities of samples using standard commercial kits306 is a potential drawback. TMT-based labelling has been applied to studying O-GalNAc84,307, O-GlcNAc308,309 and N-glycoproteomes310,311.
For sensitive applications in the clinical setting, absolute quantification of select glycopeptides is possible using internal standards such as stable isotope-labelled counterparts, which allow normalization across samples and direct comparison of analyte concentrations between different patients312,313. This approach enables reliable quantification of glycopeptides of interest in large patient cohorts, although it is limited by the time-consuming and high-cost synthesis of relevant glycopeptide standards.
Results
Comprehensive characterization of glycopeptides from MS data involves determining the peptide sequence, the site (or sites) of glycosylation and identity of the attached glycans. A growing number of software solutions enable the identification of glycosylation events (Table 3), and computational approaches associated with glycopeptide identification are rapidly developing. Below, we highlight the features of different fragmentation data and discuss the existing tools and emerging bioinformatic methods. We also highlight the conceptual frameworks that underpin glycopeptide assignments, localizing glycosylation sites and defining glycans.
Glycopeptide sequence determination
Decades of developments in proteomics have provided various robust methods for identifying peptide sequences from MS data by comparing protein sequences from a reference database in silico with the observed spectra314,315. Such methods include Mascot316, SEQUEST317, Andromeda318 and MS Amanda319. Handling the addition of attached glycans of varying complexity poses great challenges with existing proteomic workflows; below, we discuss two major approaches that address these challenges, distinguished by whether peptide fragment ions are searched with or without attached glycans.
Searching peptide ions with the attached glycan: ‘variable modification’ searches
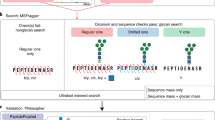
When treating attached glycans as variable modifications on peptides (Fig. 3a), possible glycan masses are specified on allowed sites, and theoretical glycopeptides containing these glycan masses are generated from the peptide sequences provided in a proteome database. The precursor mass for a given MS/MS spectrum is used to select candidate glycopeptides, which are then scored by comparing the observed MS/MS spectrum with the theoretical fragment ions of the glycopeptide candidates. Sequences supported by sufficient peptide fragment ion evidence result in a peptide spectral match (PSM). Glycopeptides present two major challenges for this approach: first, the heterogeneity of possible glycan structures can result in a huge number of candidate glycopeptides to consider when multiple possible glycosylation sites are available in a peptide sequence. Second, glycan fragments are often lost from glycopeptide ions in collisional or hybrid activation methods; as glycan modifications are specified as an integral part of the peptide in this approach, they are expected to be present in both MS1 and MS/MS spectra, and the loss of a glycan or parts thereof in the MS/MS spectrum will prevent matching theoretical ions containing the glycan (Fig. 3). For this reason, traditional proteomics tools have severely limited sensitivity for the sequencing of glycopeptides using collision-activation-based fragmentation.

a | Glycans can be searched as a variable modification of peptides, similar to how other post-translational modifications (PTMs) are identified in common proteomics searches. The in silico prediction of the search tool assumes that the fragment ions observed in the tandem mass spectrometry (MS/MS) events will preserve the glycan at the site of attachment in the peptide. b | For glycopeptides fragmented by collisional activation, offset-style searches can look for peptide ions that have lost the glycan directly within MS/MS scans. c | The glycan-first method of separating the precursor mass into peptide and glycan components uses a series of Y-type ions resulting from a known core structure to determine the glycan mass. Subtracting the glycan mass from the precursor mass yields the peptide mass, which is then used to determine candidate peptide sequences that are compared with the peptide fragment ions observed. d | The alternative peptide-first method uses an offset-style search to identify the peptide sequence from peptide fragment ions that have lost the glycans. The resulting peptide mass is subtracted from the precursor mass to yield the glycan mass, which can be matched to a specific composition or structure using the observed Y-type ions. m/z, mass to charge ratio.
Glycoproteomics-focused sequencing approaches can address the above challenges. One approach is to adapt an existing search engine to filter spectra for the presence of oxonium ions and add glycan masses to observed peptide ions112,229,320,321. A variation of this method179,322 first groups glycopeptide spectra using clustering methods before searching, allowing glycopeptide annotations to be transferred from one identified spectrum to the entire cluster. Other tools, including Byonic323,324,325,326,327, perform their own variable modification-style search with the inclusion of peptide fragment ions with various glycan additions or losses, using various scoring methods to evaluate glycopeptides (note that although this method is extremely sensitive, concerns have been raised about the accuracy of this approach328). Alternatively, tools such as Protein Prospector329 use a multi-step search, whereby an initial open search determines common glycan masses to be included in a second, more specific search330,331. Overall, variable modification searches are straightforward to implement for the localization of glycans — particularly those on glycopeptides fragmented by electron-based activation methods — although the inclusion of additional fragment types can reduce search speed, and some methods have reduced sensitivity in collision-activation data owing to glycan losses.
Searching peptide ions missing fragmented glycans: ‘offset’ searches
In offset searches, peptide sequence ions are searched directly without glycans (Fig. 3b). This offers greatly improved sensitivity over variable modification approaches for glycopeptides fragmented by collisional activation, as peptide fragments that have lost glycans (Fig. 3b) can be matched and contribute to the peptide score. The most common implementation of this method is a ‘glycan-first’ search, in which a series of Y-type ions corresponding to a common glycan core structure is used to determine the mass of the glycan and, by extension, the glycan-free peptide mass, which is then used to search for peptide fragment ions without the glycan (Fig. 3c). This approach has proved popular60,332,333,334 owing to its computational efficiency and ability to infer glycan composition information from the Y-type ions, particularly for N-glycopeptides.
In an alternative ‘peptide-first’ strategy, peptide fragment ions without glycans can be searched directly in the MS/MS spectra using an open or mass-offset search (Fig. 3d). These searches335,336 use computational advances to allow peptide fragment ions to be matched in MS/MS spectra even if the peptide sequence mass does not match the observed molecular mass. This approach eliminates the need to match a Y-type ion series, providing a sensitivity boost for glycopeptides that carry labile glycans or do not produce prominent Y-type ions.
Finally, spectral library methods, such as those used for DIA-based analysis, circumvent the need for glycan-first or peptide-first searching by matching observed MS/MS signals to annotated glycopeptide fragmentation spectra286,337,338,339. This technique gives sensitive quantification at the cost of requiring a separate analysis to build the spectral library and limiting identifications to glycopeptides present in the library.
Glycosylation site localization
Methods for locating the site or sites of glycan attachments in a peptide are varied depending on the type of glycosylation being considered. For example, most tryptic N-glycopeptides have only a single possible glycosylation site corresponding to the consensus sequon asparagine-X-serine/threonine (where X can be any amino acid except proline). The predictable nature of N-linked glycosylation allows the inference of glycan location, often without the need for additional spectral evidence. In peptides with multiple sequons or combinations of glycosylation types, N-glycans can be localized directly using ExD or hybrid-type activation and searching for intact glycans with variable modification-style methods340,341 or peptide fragment ions retaining a glycan remnant using collisional activation252,335.
Experimental localization of O-glycosylation sites on peptides represents an important yet challenging task owing to the lack of a universal deglycosylation enzyme effective for all O-glycan core structures. This prevents the application of ‘de-glycoproteomic’ approaches common to N-glycan site localization, in which N-glycans are removed by PNGase F, allowing glycan sites to be determined by identifying deamidated residues within an N-glycosylation sequon. Localization of O-glycans is complicated by the lack of a consensus sequon to reduce the number of possible glycosylation sites in a peptide, their facile dissociation from the peptide carrier upon collisional activation and the high density of occupied O-glycosylation sites on peptides from mucin and mucin-domain glycoproteins. Therefore, O-glycosite localization requires the analysis of intact glycopeptides using electron-based or hybrid-type activation methods, which produce peptide fragment ions that preserve glycan conjugation252. In favourable cases, such as highly charged glycopeptides, variable modification-style searches can provide high-confidence O-site localization from electron-based activation329. Peptides with multiple possible glycosylation can have a huge number of potential glycan configurations, and, as a result, most variable modification-style searches are restricted to only the most commonly occurring glycans. To address this combinatorial limitation, open or mass-offset search methods first identify the peptide sequence and total glycan mass, reducing the search space to allow the localization of individual glycans. Protein Prospector performs such a multi-step search for electron-driven fragmentation329. The O-Pair search introduced in MetaMorpheus336 and a similar method implemented in pGlyco3 (ref.342) use paired collisional and electron-based ion activation scans, performing a mass-offset search of the collisional scan to identify the glycopeptide sequence and total glycan mass, followed by dynamic programming to decompose the total glycan mass into multiple individual glycans and localize each within the peptide (Table 3). This highly promising approach takes advantage of the sensitivity of offset searches and collisional activation to identify glycopeptides and the ability of electron-based activation to localize glycosites.
Glycan identification
Paired glycomic and proteomic analyses of PNGase F-treated samples can provide detailed characterization of glycans and deglycosylated glycosites310,343. Glycomics provides useful (and still unmatched) structural insight into the protein-linked glycans in a protein mixture; however, undertaking parallel proteomics and glycomics workflows is time-consuming and reduces overall sensitivity. This has prompted the development of methods that can characterize some glycan structural features directly from intact glycopeptides. The determination of the monosaccharide composition of glycans is complicated by the multiple isomeric and isobaric compositions and structures possible for an observed molecular mass62,343, an analytical challenge exacerbated by the existence of common peptide modifications such as oxidation, deamidation and carbamidomethylation that mimic the mass difference between different glycan compositions61. Compositions can in most cases be discriminated using glycan fragment ions, similarly to how a peptide is sequenced using peptide fragment ions. Collisional fragmentation energies that generate glycan fragment ions are often lower than those optimal for peptide backbone fragmentation, creating a trade-off between optimizing glycan and peptide fragmentation in the LC–MS/MS experiment. SCE-beamCID and paired low-energy and high-energy beamCID experiments have shown great promise in this area55,344.
Many published studies of intact glycopeptides report only the mass of a glycan, or a putative composition or structure, assuming that there is only a single composition or structure for the detected glycan mass. This approach, used by many tools229,327,332,336,338,345 (software tools are listed in Table 3) greatly simplifies data handling; however, it does not consider isomeric glycans, which may have biological implications. As the existence of multiple isomeric glycans is often not known in advance, this can potentially result in incorrect assignments when a single form is assumed.
Several methods to assign glycan compositions and/or structures directly from glycopeptide fragmentation data have been developed recently. Glycan-first offset searches are a natural fit for these approaches given their reliance on the identification of Y-type ions, with several programs implementing glycan assignments with various scoring methods60,333,334,337,346,347. The peptide-first glyco search in MSFragger can perform a combined Y-type ion and oxonium-ion composition assignment method as a post-processing step348. Compared with the glycan-first approach, in which all glycans are scored against each spectrum, the peptide-first approach greatly simplifies glycan assignment as the glycan mass is known before assignment, making it easier to distinguish between glycans with similar or identical masses. Finally, variable modification searches have been demonstrated using Y-type ions to distinguish glycan compositions using a database of possible glycans or de novo from a range of possible glycans320,323,324,325,326,331.
Stereochemical and positional glycan information can be extracted from glycopeptide MS/MS information38,349 (Table 4). For example, ratios of specific oxonium ions can be used to discriminate between glycopeptides bearing isomeric O-GlcNAc and O-GalNAc moieties350 and are also useful for crude classification of N-glycopeptides versus O-glycopeptides252,351. Ratios of oxonium and B-type ions can also distinguish between α2,3-sialyl and α2,6-sialyl linkages352, between core (α1,6-) and antenna (α1,2/3/4-) fucosylation248,255,344, and between some classes of mucin-type O-glycosylation266,272,353,354. Y-type ions generated at low beamCID energies can also be used for determination of core fucosylation255,355,356, bisecting GlcNAc-containing glycopeptides248,357, and various antennary structures272,337,353,358,359. Furthermore, oxonium ions specific to chemical groups introduced by glycan labelling can be useful for structural characterization171,172,360,361,362,363. Although many of these diagnostic ions occur through beamCID fragmentation, they can also be observed in hybrid ExD spectra such as those collected by EThcD. Despite the promise that diagnostic ions provide for structural characterization, a major challenge is the co-elution of glycoforms (see below). Without chromatographic, electrophoretic or mobility separations of related glycoforms, diagnostic ions characteristic of multiple structures may be present in a single MS/MS spectrum.
Statistical control of assignments
Controlling the FDR for peptide sequence assignment has received considerable attention. FDR methods for peptide sequence assignment involve generating decoy peptides by reversing or scrambling target peptide amino acid sequences and using the ratio of decoy-to-target peptide matches to estimate the score threshold required to achieve a given FDR364. Within glycoproteomic studies, FDR methods based solely on peptide sequence determination have been suggested to provide partial correct FDR control, although multiple groups have highlighted higher-than-anticipated FDRs in glycopeptide data sets62,273. Attempts to overcome inadequate FDR controls include additional score cut-offs to limit potentially erroneous assignments57,62,252 and manual inspection of glycopeptides76,77. Further, computational approaches have been proposed to control glycopeptide FDRs at both the glycan and peptide levels54,348.
In contrast to the statistical controls for the peptide sequences assigned to glycopeptides, which are generally considered robust, the determination of glycan composition or structure is acknowledged to be a key limitation of intact glycopeptide analysis365. The software tools for the determination of glycan composition described above use a fragment-ion-based method for assigning glycans, and the accuracy of such assignments has largely been evaluated manually or with empirically determined score filters62. Manual expert-based curation of output data is time-consuming and often prohibitive for large-scale analysis of glycopeptides, prompting the development of glycan-specific FDR methods to enable automated control of false assignments. The linear sequence of amino acid residues can be reversed or shuffled to make a decoy peptide with the same amino acid composition as the target; however, non-linear glycans comprising multiple different building blocks of identical masses require a different method for decoy generation. GlycoPepEvaluator366 and IQ-GPA323 generate decoys by substituting monosaccharides and reversing or altering the glycopeptide sequence to obtain a decoy glycopeptide that is an isobar of the target glycopeptide and that contains a nonsensical glycan (Table 3). An alternative ‘spectrum-based’ FDR method implemented in GlycoPAT324 and pGlyco346 generates decoy glycans by applying random mass shifts to the fragment ions of a target glycan, preserving the fragmentation characteristics of the target glycan and assessing the likelihood of random matches to ions in the mass spectrum. This approach has been adopted by GPSeeker60, GlycReSoft177,325, MSFragger glyco335 and StrucGP347 (Table 3). Care must be exercised using these techniques as the provided FDRs may not hold when faced with unexpected glycans not present in the provided database, or oxonium ions resulting from co-fragmentation of co-eluting isobaric glycopeptides.
Once identified, statistical assessments are also applied to identify quantitative changes in glycopeptides such as Student’s t tests, which are commonly used for comparisons between binary conditions52. Multiple sample comparison approaches such as ANOVA are also widely implemented if multiple groups are to be compared310,311. For these comparisons at least a onefold change in abundance is typically required to be considered a change and the P value threshold should be tailored to the experiment using multiple hypothesis corrections to ensure further confidence in the observed changes52,310,311. Threshold-based approaches are typically favoured for studies investigating the substrates of specific glycotransferases; where glycopeptides with 10-fold79,81 or 100-fold158 changes in the absence of the glycotransferase in question are considered as potential substrates. Changes observed at the glycosylation level can be driven by both changes in glycosylation occupancy and changes in the total protein level, and normalization against proteomics data can therefore be advantageous79,81,158,293.
Applications
Glycoproteomics has a range of applications in the clinical sciences. The study of complex biological samples from clinically relevant specimens such as tissue biopsy samples, blood, urine and cerebrospinal fluid (CSF) has provided an opportunity to understand the fundamental roles of glycosylation in pathophysiology. Furthermore, glycoproteomics has aided the search for diagnostic and prognostic biomarkers that can stratify patients for specific interventions and follow disease progression. Most of these biomarker studies have aimed to identify and quantify glycopeptides and glycoproteins or determine the occupancy of specific glycosites to identify differential changes in protein glycosylation patterns in conditions of health and disease (Fig. 4). Additionally, glycoproteomic data are increasingly being combined with data from other omic methods such as transcriptomics, proteomics, glycomics, phosphoproteomics and metabolomics52,83,367 to better understand the connection between site-specific glycosylation and the various biological processes that take place in complex systems. So far, most glycoproteomic studies that incorporate multi-omics have focused on N-glycoproteomics, although there are also a few examples for O-glycoproteomics as discussed below.

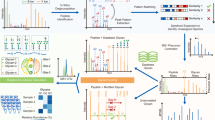
a | Bottom-up glycoproteomic studies using clinical samples from healthy controls and patients (in this hypothetical case, cerebrospinal fluid (CSF) from healthy controls and patients with Alzheimer disease (AD)) can identify prognostic and diagnostic biomarkers through finding glycopeptides that are differentially regulated between the two populations. Top: examples of glycopeptides not differentially regulated by disease conditions. Middle: differentially regulated glycopeptides. Bottom: loss of glycosylation in disease conditions. Dashed boxes indicate selected biomarker candidates. Volcano plots such as that displayed can show significant differences in abundance of glycopeptides from control and patient samples. Volcano plot generated using the VolcaNoseR online resource. b | Following the discovery of candidate biomarkers, larger patient cohorts can be used to validate selected glycopeptides by targeted parallel reaction monitoring (PRM) and liquid chromatography–mass spectrometry (LC–MS) to monitor specific glycopeptides of interest across control and patient-derived samples. These studies, which can focus on the identification of specific glycoforms (biomarker 1) or the absence or presence of glycosylation events (biomarker 2), aim to confirm that the markers of interest enable the separation of groups, such as a control group (CTRL) from an AD cohort at a population level. c | Standardized assays can be developed for validated candidates to aid in diagnosis. For example, specific changes in the predominant glycosylation of an isolated protein or peptide can be detected using a lectin-based enzyme-linked immunosorbent assay (ELISA) (left panel). Alternatively, loss of glycosylation can be pursued through targeted PRM analysis, in which spiking known amounts of a stable isotope-labelled peptide counterpart allows direct comparison of analyte amounts in different clinical specimens (right panel). Combining such biomarkers can lead to improved diagnostic and prognostic characteristics. AUC, area under the curve; m/z, mass to charge ratio; ROC, receiver operating characteristic; RT, retention time.
Mapping N-glycosylation for diagnostics
Many studies have mapped N-glycosites in patient-derived biofluids or cellular material to identify informative biomarkers for diagnostic and prognostic applications368,369. N-glycoproteomics has been extensively used to analyse various sources of neural tissue in an attempt to identify biomarkers for neural diseases, including stem cell-derived neural cells, mouse brains and patient-derived CSF88,370,371. Recently, comparative in-depth N-glycoproteomic analysis of CSF samples from healthy controls and patients with Alzheimer disease demonstrated differential N-glycosylation patterns between cohorts368. Similarly, comparisons of postmortem human Alzheimer disease and control brain tissue have shown quantitative changes in N-glycosite occupancy in clinically relevant proteins372.
N-glycoproteomics has also been explored as a tool for the early detection of cancer. Cancer models studied so far include ovarian cancer cell lines with differential resistance to the chemotherapeutic agent doxorubicin373, as well as patient serum samples369, and native and xenografted tissues from ovarian serous carcinoma369,374,375. These studies have demonstrated that the detection of select glycopeptide signatures may be useful in diagnostic applications, for the stratification of patients or to follow disease progression. Studies in other cancers have also shown differential abundance of select N-glycopeptides between tissues, serum and bodily fluids from healthy donors and patients with cancer, further suggesting that alterations in specific N-linked glycosylation events may correlate with cancer progression13,376 and that the integration of N-glycoproteomic profiles can improve diagnostic sensitivity compared with proteomics alone377,378,379,380,381.
Mapping O-glycosylation
The application of O-glycoproteomics to a range of biological questions has resulted in a massive expansion of the mammalian O-glycoproteome53,76,77,124, leading to unexpected discoveries such as the discovery of O-glycosylated neuropeptides and peptide hormones67,382, O-glycans in LDLR-related protein linker sequences80 and extensive O-glycosylation of viral envelope proteins66,78.
The discovery of O-glycoproteases and their inactive mutants has led to the development of O-glycoprotein and mucin-domain glycoprotein enrichment methods. A notable example of using catalytically active O-glycoproteases for O-glycosite enrichment is the site-specific extraction of O-linked glycopeptides (ExoO) approach, which has been used to identify O-glycosites on more than 1,000 proteins across human kidney tissue, T cells and serum samples124. Inactive O-glycoproteases have also been shown to be robust affinity tools for enabling the differentiation of cancer-associated changes in mucin-domain-containing glycoproteins96,125. A recent preprint publication showed that inactive StcE-based enrichment was capable of isolating hundreds of O-glycopeptides from patient-derived ascites fluid, including many from MUC16 — the classic, gold-standard biomarker for ovarian cancer383.
Genetic knockouts of specific GalNAc-Ts have identified isoform-specific substrates in various cell lines and tissues68,79,81,82,83 that could give information on the pathophysiological mechanisms that drive congenital disorders of glycosylation384. Further genetic engineering-driven glycoproteomic strategies — first using zinc finger nucleases76,385 and more recently CRISPR-based approaches83,158 — have led to the discovery of novel glycosylation pathways such as an O-mannosylation system responsible for glycosylation of cadherins158. These discovery-driven applications of glycoproteomics have expanded our understanding of carbohydrate-binding proteins386,387,388, providing insights into how glycan recognition may have an important role in cancer development.
Glycoproteomics in multi-omic studies
Multi-omic approaches that combine transcriptomic and glycoproteomic analyses can provide context for the global consequences of N-glycoproteomic or O-glycoproteomic changes in cell systems, disease models and clinical specimens79,115,116,389,390. For example, in a clinical setting, combining N-glycoproteomic-based classification of tumours with transcriptomic changes led to biomarker discovery and prospective therapeutic targets based on the pathways identified391. Further, public genomic, transcriptomic or proteomic repositories of patient cohort data can be excellent sources of data for correlation with glycoproteomic data381, an approach that has been used to help understand global regulatory networks in cell differentiation programmes392.
Another successful multi-omic approach is to combine glycoproteomic data with data from phosphoproteomic analyses393,394,395,396,397. The integration of phosphoproteomics, proteomics, transcriptomics and glycoproteomics can provide comprehensive insights into disease mechanisms or tissue development, as recently shown for both N-linked and O-linked glycans83,398. In such multi-omic studies, transcript expression can be correlated with protein expression, and cross-referencing of PTMs with protein abundance and signalling networks gives a narrow selection of relevant targets for downstream study83.
The modelling of glycans at specific sites can be useful for understanding the functional impacts of changes in glycosylation. Multiple platforms provide tools for predicting 3D structures of carbohydrates attached to glycoproteins399, and it has been argued that new tools such as AlphaFold2 should be modifiable to incorporate PTMs such as glycosylation, which will enable far more realistic structural predictions400. Integrative bioinformatics tools such as the GlycoDomainViewer401 are now also beginning to emerge, which allows glycosylation sites to be assessed within the context of the protein sequence, domain architecture and other known PTM events.
Although MS-based glycoproteomic applications are becoming more mainstream, several challenges remain. Comprehensive characterization of glycosite microheterogeneity and reliable quantification of glycopeptides harbouring different glycans is still challenging in complex clinical samples. These challenges are exacerbated when the amount of sample is limited and when multi-omic analysis from an identical sample is required. Methods that preserve the natural context and provide reliable quantification should be prioritized given the limitations of cell culture-based systems. One of the next milestones for the community will be applying glycoproteomics at the level of individual cell types, or even at the single-cell level, which could provide insight into the spatiotemporal regulation of glycosylation in different tissues. Recent progress in MOE labelling has now shown that cell line-specific glycoprotein tagging can be achieved within in vivo models (as shown in a recent preprint article), opening new opportunities to explore cell lineage glycoproteomes in native contexts402. As the field develops, translating the findings of glycosite mapping studies into a deeper understanding of the molecular mechanisms regulated by glycosylation will become the central goal of glycoproteomics.
Reproducibility and data deposition
Glycoproteomics is still a maturing field and, unlike proteomics and other omic disciplines, has yet to experience consolidation and harmonization of its experimental methodologies and informatics approaches. As the glycoproteomics community grows, it will be important to establish conventions and move towards the use of standardized approaches that reflect best practice for the collection, management and sharing of data. Below, we discuss factors that lead to known reproducibility issues.
Variations in data collection
A key factor that contributes to the lack of reproducibility in glycopeptide data sets across laboratories is the inconsistent and often incomplete description of sample handling, sample processing and data acquisition parameters such as those relating to LC–MS/MS experiments. Experimental variations in peptide generation, chemical derivatization or labelling steps and glycopeptide enrichment can greatly affect the resulting glycopeptide data and are often not fully explained. These differences can be compounded in the LC–MS/MS acquisition process by, for example, changing MS ionization and fragmentation behaviours. For these reasons, it is crucial to fully describe these parameters in published research. It should be noted that MS instrument cleanliness and chromatography performance are also vitally important for data integrity403.
A diverse set of experimental methods are available for glycoproteomics data generation as demonstrated by several glycopeptide-focused multi-laboratory studies conducted through the Human Proteome Organization’s Human Disease Glycomics/Proteome Initiative404,405, the Association of Biomolecular Resource Facilities406 and the National Institute of Standards and Technology (NIST)407. Although analytical diversity could be considered a strength of the field, several of these experimental methods, some using highly customized and non-commercial reagents, are employed by few groups worldwide, therefore making data difficult to reproduce. Standardization of methods across laboratories could reduce some of these observed variations, although we acknowledge it is unlikely that a one-size-fits-all approach to methodologies would be advantageous for many biological questions.
Variations in data analysis
Analysis of glycopeptide data is challenging and a source of variation in glycoproteomic experiments. A recent multi-institutional study performed by the Human Proteome Organization (HUPO) Human Glycoproteomics Initiative evaluating software tools for serum N-glycopeptide and O-glycopeptide analysis using glycopeptide data sets provided from various glycoproteomic laboratories found that the identified glycopeptides varied dramatically between laboratories even when the same informatic tools were employed, confirming that variables such as pre-processing and post-processing methods substantially affect glycopeptide assignments even on identical data sets365. Although this comparison identified several high-performance search strategies, the large variability in the performance of software tools and search parameters highlights that ongoing benchmarking to track and compare the performance of glycoproteomic informatics used across the community is crucial.
Data deposition and sharing
Data repositories will be essential for glycoproteomics data to comply with the FAIR data deposition standards408. The MIRAGE initiative has taken the lead in proposing reporting guidelines for glycomics409, and these are currently undergoing refinement to provide guidelines for glycoproteomic data. The MIRAGE guidelines have been adopted by several journals to ensure that consistent information is reported for glycomic experiments with the goal that the finalized glycoproteomics guidelines will provide a clear framework for the glycoproteomics community. To facilitate the sharing of data, glycoproteomic-centric repositories have been launched, for example, GlycoPOST410, which assigns unique identifiers to raw MS data for individual projects and provides input forms and spreadsheets to give users a template for providing metadata required by MIRAGE guidelines. The database UniCarb-DR411 is complementary to GlycoPOST and allows users to visualize glycan structures annotated in the raw MS data. At the time of writing, UniCarb-DR and GlycoPOST are both available from the GlyCosmos Glycoscience Portal412. ProteomeXchange413,414 is also available for (glyco)proteomic LC–MS data deposition. As avenues for data sharing are now established, all published glycoproteomic data should be made publicly available. Many journals are already beginning to implement this requirement and it is important to note that ensuring the public availability of data will be a community effort.
Limitations and optimizations
Several assumptions and experimental trade-offs shape the conclusions that can be drawn from glycoproteomic studies. Although workflows used to undertake glycoproteomics are continuously improving, a clear understanding of potential limitations and the underpinning assumptions associated with these workflows is needed to best interpret glycoproteomic data.
One MS/MS event, multiple glycoforms
A common assumption for glycopeptide MS/MS events is that each of the resulting spectra contains a single glycoform; however, multiple isobaric glycans57 or isomeric glycosylation states415 may be observed within a single MS/MS spectrum. Isobaric glycans and isomeric glycopeptides possess similar elution profiles when separated using chromatography approaches such as RP-LC, resulting in mixtures of glycoforms being subjected to MS/MS analysis (Fig. 5a). This leads to the generation of chimeric spectra that complicate the assignment of glycosylation sites and glycan arrangements (Fig. 5). Chimeric spectra have been observed in N-linked glycoproteomic studies54, and O-linked glycopeptides are known to display multiple isomeric species63. Careful analysis of chromatography-separated isomers270 or use of additional separation techniques such as IMS216 can help to resolve co-eluting isomeric glycosylation sites.

Glycopeptide co-elution and co-isolation of isomeric species can lead to the generation of chimeric spectra containing fragments from two or more precursor ions. a | Glycopeptide isomers can possess unique elution properties when separated with reverse phase separation, although some isomers may have closely related elution profiles. b | The presence of multiple glycopeptide isomers in samples can result in the observation of multiple overlapping Gaussian features in the chromatogram. c,d | Examining tandem mass spectrometry (MS/MS) spectra corresponding to different retention times results in distinct MS/MS spectra containing different mixtures of isomeric glycopeptide species. These chimeric spectra are identifiable by the presence of fragment ions corresponding to the modification attached to two residues, such as the c12 and z4 ions highlighted in blue. Mixtures of isomeric glycopeptides can result in chimeric spectra, supporting the assignment of mutually exclusive glycosylation events. ETD, electron transfer dissociation; m/z, mass to charge ratio.
MS-based glycan class assignments
MS data provide limited insights into monosaccharide identity or linkage information (see above). This lack of information limits the ability to assign glycan classes on the basis of mass alone. Although the conservation of glycosylation pathways in eukaryotic glycosylation systems does constrain many glycan compositions, which allows glycan classes to be predicted and/or assigned with reasonable confidence416,417, it is important to note that these should still be treated as unconfirmed assignments. Orthogonal methodologies can be used to further support the presence of specific glycans or linkage configurations such as the use of exoglycosidases418; the release of glycans and confirmation of specific glycans using isomeric resolving approaches such as PGC310,367; or the analysis of oxonium fragmentation patterns to support monosaccharide assignments114,350. In situations where glycans are ambiguous, restraint in the assignments of glycan classes is best practice. Alternatively, an increasingly accessible way to corroborate glycopeptide assignments is the use of synthetic glycopeptide standards, which allow subtle changes in retention time or fragmentation properties to be detected to support glycan identities419.
Ambiguous localizations
The community’s ability to assign glycosylation sites has seen a dramatic improvement over the past decade with multiple innovations in instrumentation and data acquisition, such as increased accessibility to ExD dissociation methods on multiple instrument platforms and improved data collection approaches57,341. These innovations do not guarantee that localization information will be obtained for a given glycopeptide, and a large proportion of glycopeptides are not able to be localized within most data sets. A growing question within the field is whether site localization is needed for all glycosylation experiments, especially if localization comes at the cost of speed and subsequent glycoproteomic depth252. Glycopeptide-focused DIA analysis286,339, which is undertaken using beamCID, highlights this change in thinking and the growing acceptance of site ambiguity. Many in the field advocate that sites should be assigned either as localized or non-localized on the basis of the available fragmentation information76,77,286 (Box 2). Further, a formal system to stratify glycosylation site ambiguity on the basis of site localization probability was recently proposed by Lu, Riley et al.336 to provide a means to categorize assignment quality. In reality, not all biological questions need complete unambiguous glycosylation site assignments; for example, studies in which the focus is the identification of glycans367,420 or the quantification of glycopeptide abundances52,310 will not be affected by site ambiguity. By contrast, site localization can be crucial for confirming atypical glycosylation events such as tyrosine O-glycosylation76,421 or when attempting to fully characterize the site-specific glycosylation of a protein of interest, especially when both N-glycans and O-glycans are present. It should be noted that at least partial localization of glycans may be required for peptides with multiple glycosylations to avoid misassignment of glycan compositions63,335.

Outlook
Glycosylation shapes nearly all biological processes across all areas of life, and there has been a rapid growth in glycobiology-focused efforts over the past two decades to define and understand the role of the complex and dynamic glycoproteome. The development of chemical biology tools for tagging glycoproteins93,162,169,170, enrichment techniques to isolate glycopeptides114,129 and new glycoproteome-specific reagents such as O-glycoproteases96,125 have greatly improved our ability to site-specifically map glycosylation across biological systems. Over the coming years, improved access to glycoproteomics toolkits promises to stimulate further activity in the field and promote an increasing number of studies exploring fundamental and applied questions in glycobiology.
Glycoproteomics has shown potential to differentiate disease subtypes, stratify patients and predict clinical outcomes in complex human diseases such as cancer398,422, inflammation423,424 and microbial infections425,426, and there is great potential for glycoproteomic analysis to improve diagnostic sensitivity and precision376. Community-based development of robust methods and software that implement best practice for data interpretation, standardization and sharing will be essential for clinical translation; this has begun with the establishment of glycoproteomic focused sharing platforms such as GlycoPOST410. Although these developments are promising, ease of use and implementation is still the major hurdle currently limiting the translation of glycoproteomics to the clinic.
It is important for glycopeptide-focused software solutions to be developed in parallel with new practical techniques. Future tools should aim to be customizable to facilitate the analysis of diverse glycoproteomes beyond the mammalian realm, including in plants, invertebrates and microbial systems31,33,34. For future software solutions, the crucial challenges will be identifying and localizing multi-glycosylated peptides, statistical control of glycopeptide identification and distinguishing glycan structural isomers.
Marked improvements in proteomic sample multiplexing, chromatography and MS acquisition speed are likely to lead to increased throughput in the field of glycoproteomics. Peptide-based sample multiplexing techniques using tandem mass tags currently allow 18 samples to be analysed within a single proteomic experiment304. Multiplexing can also be used to provide structural insights by allowing the incorporation of samples treated with specific glycosylation inhibitors112 or the inclusion of genetic knockouts of specific glycosyltransferases or glycoside hydrolases371,427, enabling glycan class or isoform information to be obtained that may otherwise be missed.
Improvements in glycoproteomic depth are likely to come from new tools. The recent demonstration of a large range of bacterial glycan-targeting hydrolytic enzymes125 shows that the current repertoire of glycoproteases represents only a small subset of possible enzymatic activities and specificities. As our understanding of glycan-modifying enzymes improves428,429, so too will our ability to rationally modify and tailor these enzymes to target or enrich specific glycosylation sites and their glycans of interest. Modified enzymes and affinity tools generated against specific glycans430 will be particularly valuable to advance less-mature areas of glycoproteomics such as C-glycosylation431. Additional methods for unbiased, untargeted quantitative profiling of multiple glycosylation classes in a single experiment will also be crucial.
Applications such as single-cell analysis and top-down glycoproteomics still represent significant technical barriers for the field. Although isobaric labelling approaches are increasingly used for single-cell proteomic analysis432,433 and have the potential to enable single-cell glycoproteomics, it remains to be seen how applicable these approaches will be. The use of charge detection MS434,435,436 has the potential to radically improve top-down glycoform characterization, and integration of these approaches for glycoproteomics will require further development. Non-MS-based DNA-sequencing methods using oligonucleotide-labelled lectins have been used by several groups to explore glycosylation changes at the single-cell level113,437. Further, a recent study demonstrated that non-glycosylated and glycosylated forms of peptides can be resolved using nanopore sequencing438, suggesting that this technique may enable single-molecule analysis of glycopeptides and glycoproteins. Although these technologies are still in their infancy, they have considerable potential to provide orthogonal information to MS-based glycoproteomics.
Great strides have been made in glycoproteomics-based identification of glycosylation events and the discovery of new or unusual types of protein glycosylation33,367,439. Over the coming years, glycoproteomics will increasingly provide valuable mechanistic insight into the formation and role of protein-linked glycans in biological processes. New insights into mechanisms such as the requirement of N-linked fucosylation for ricin toxicity371 or the role of specific O-GlcNAcylation sites in metabolic regulation440 have already been established using glycoproteomics. Further, multi-omic integration has enabled a holistic understanding of biological systems and it is likely that the integration of glycoproteomics with other omic techniques for the analysis of large cohorts will further enhance our knowledge at a population level. For example, the identification of common genetic variants associated with differences in glycosylation through genome-wide association studies may further enhance mechanistic insights and unravel potential disease predispositions424,441.
As methods and technologies continue to evolve, one of the most exciting opportunities for the field will be further integration and improvements in the bioinformatic space. Across the life sciences, the growing application of machine learning approaches is leading to new ways to model, analyse and handle large data sets of increasing complexity and information content442,443. Machine learning and artificial intelligence are not used routinely by the glycoproteomics community, although their increasing use in proteomics444 suggests that these approaches will become commonplace in glycoproteomics workflows. Collectively, these transformative tools are likely to make glycosylation analysis accessible to a wider range of life scientists, ultimately improving our understanding of organismal development, disease adaptation and evolution.
References
Freeze, H. H., Eklund, E. A., Ng, B. G. & Patterson, M. C. Neurological aspects of human glycosylation disorders. Annu. Rev. Neurosci. 38, 105–125 (2015).
Ohtsubo, K. & Marth, J. D. Glycosylation in cellular mechanisms of health and disease. Cell 126, 855–867 (2006).
Varki, A. Biological roles of glycans. Glycobiology 27, 3–49 (2017).
Stowell, S. R., Ju, T. & Cummings, R. D. Protein glycosylation in cancer. Annu. Rev. Pathol. 10, 473–510 (2015).
Reily, C., Stewart, T. J., Renfrow, M. B. & Novak, J. Glycosylation in health and disease. Nat. Rev. Nephrol. 15, 346–366 (2019).
Wandall, H. H., Nielsen, M. A. I., King-Smith, S., de Haan, N. & Bagdonaite, I. Global functions of O-glycosylation: promises and challenges in O-glycobiology. FEBS J. 288, 7183–7212 (2021).
Tran, D. T. & Ten Hagen, K. G. Mucin-type O-glycosylation during development. J. Biol. Chem. 288, 6921–6929 (2013).
Kaltner, H., Abad-Rodriguez, J., Corfield, A. P., Kopitz, J. & Gabius, H. J. The sugar code: letters and vocabulary, writers, editors and readers and biosignificance of functional glycan-lectin pairing. Biochem. J. 476, 2623–2655 (2019).
Lin, B., Qing, X., Liao, J. & Zhuo, K. Role of protein glycosylation in host-pathogen interaction. Cells 9, 1022 (2020).
Thompson, A. J., de Vries, R. P. & Paulson, J. C. Virus recognition of glycan receptors. Curr. Opin. Virol. 34, 117–129 (2019).
Pereira, M. S. et al. Glycans as key checkpoints of T cell activity and function. Front. Immunol. 9, 2754 (2018).
Ugonotti, J., Chatterjee, S. & Thaysen-Andersen, M. Structural and functional diversity of neutrophil glycosylation in innate immunity and related disorders. Mol. Asp. Med. 79, 100882 (2021).
Rodrigues, J. G. et al. Glycosylation in cancer: selected roles in tumour progression, immune modulation and metastasis. Cell Immunol. 333, 46–57 (2018).
Zhang, L. & Ten Hagen, K. G. Pleiotropic effects of O-glycosylation in colon cancer. J. Biol. Chem. 293, 1315–1316 (2018).
Chatterjee, S. et al. Protein paucimannosylation is an enriched N-glycosylation signature of human cancers. Proteomics 19, e1900010 (2019).
Chatterjee, S. et al. Trends in oligomannosylation and alpha1,2-mannosidase expression in human cancers. Oncotarget 12, 2188–2205 (2021).
Loke, I., Kolarich, D., Packer, N. H. & Thaysen-Andersen, M. Emerging roles of protein mannosylation in inflammation and infection. Mol. Asp. Med. 51, 31–55 (2016).
Bhat, A. H., Maity, S., Giri, K. & Ambatipudi, K. Protein glycosylation: sweet or bitter for bacterial pathogens? Crit. Rev. Microbiol. 45, 82–102 (2019).
Hare, N. J. et al. Mycobacterium tuberculosis infection manipulates the glycosylation machinery and the N-glycoproteome of human macrophages and their microparticles. J. Proteome Res. 16, 247–263 (2017).
Delannoy, C. et al. Mycobacterium bovis BCG infection alters the macrophage N-glycome. Mol. Omics 16, 345–354 (2020).
Chatterjee, S. et al. Serum N-glycomics stratifies bacteremic patients infected with different pathogens. J. Clin. Med. 10, 516 (2021).
Groux-Degroote, S., Cavdarli, S., Uchimura, K., Allain, F. & Delannoy, P. Glycosylation changes in inflammatory diseases. Adv. Protein Chem. Struct. Biol. 119, 111–156 (2020).
Mealer, R. G. et al. Glycobiology and schizophrenia: a biological hypothesis emerging from genomic research. Mol. Psychiatry 25, 3129–3139 (2020).
Paprocka, J., Jezela-Stanek, A., Tylki-Szymanska, A. & Grunewald, S. Congenital disorders of glycosylation from a neurological perspective. Brain Sci 11, 88 (2021).
Ondruskova, N., Cechova, A., Hansikova, H., Honzik, T. & Jaeken, J. Congenital disorders of glycosylation: Still “hot” in 2020. Biochim. Biophys. Acta Gen. Subj. 1865, 129751 (2021).
Stambuk, T., Klasic, M., Zoldos, V. & Lauc, G. N-glycans as functional effectors of genetic and epigenetic disease risk. Mol. Asp. Med. 79, 100891 (2021).
Axford, J. et al. Translational glycobiology: from bench to bedside. J. R. Soc. Med. 112, 424–427 (2019).
Chang, D. & Zaia, J. Why glycosylation matters in building a better flu vaccine. Mol. Cell Proteom. 18, 2348–2358 (2019).
Tjondro, H. C., Loke, I., Chatterjee, S. & Thaysen-Andersen, M. Human protein paucimannosylation: cues from the eukaryotic kingdoms. Biol. Rev. Camb. Philos. Soc. 94, 2068–2100 (2019).
Schaffer, C. & Messner, P. Emerging facets of prokaryotic glycosylation. FEMS Microbiol. Rev. 41, 49–91 (2017).
Eichler, J. Extreme sweetness: protein glycosylation in Archaea. Nat. Rev. Microbiol. 11, 151–156 (2013).
Schjoldager, K. T., Narimatsu, Y., Joshi, H. J. & Clausen, H. Global view of human protein glycosylation pathways and functions. Nat. Rev. Mol. Cell Biol. 21, 729–749 (2020).
West, C. M., Malzl, D., Hykollari, A. & Wilson, I. B. H. Glycomics, glycoproteomics, and glycogenomics: an inter-taxa evolutionary perspective. Mol. Cell. Proteom. 20, 100024 (2021).
Koomey, M. O-linked protein glycosylation in bacteria: snapshots and current perspectives. Curr. Opin. Struct. Biol. 56, 198–203 (2019).
Thaysen-Andersen, M., Packer, N. H. & Schulz, B. L. Maturing glycoproteomics technologies provide unique structural insights into the N-glycoproteome and its regulation in health and disease. Mol. Cell. Proteom. 15, 1773–1790 (2016).
Levery, S. B. et al. Advances in mass spectrometry driven O-glycoproteomics. Biochim. Biophys. Acta 1850, 33–42 (2015).
Thomas, D. R. & Scott, N. E. Glycoproteomics: growing up fast. Curr. Opin. Struct. Biol. 68, 18–25 (2020).
Chernykh, A., Kawahara, R. & Thaysen-Andersen, M. Towards structure-focused glycoproteomics. Biochem. Soc. Trans. 49, 161–186 (2021).
Lageveen-Kammeijer, G. S. M., Kuster, B., Reusch, D. & Wuhrer, M. High sensitivity glycomics in biomedicine. Mass Spectrom. Rev. https://doi.org/10.1002/mas.21730 (2021).
Sun, S. et al. N-GlycositeAtlas: a database resource for mass spectrometry-based human N-linked glycoprotein and glycosylation site mapping. Clin. Proteom. 16, 35 (2019).
Thaysen-Andersen, M. & Packer, N. H. Advances in LC-MS/MS-based glycoproteomics: getting closer to system-wide site-specific mapping of the N- and O-glycoproteome. Biochim. Biophys. Acta 1844, 1437–1452 (2014).
Darula, Z. & Medzihradszky, K. F. Analysis of mammalian O-glycopeptides-we have made a good start, but there is a long way to go. Mol. Cell Proteom. 17, 2–17 (2018).
Khoo, K. H. Advances toward mapping the full extent of protein site-specific O-GalNAc glycosylation that better reflects underlying glycomic complexity. Curr. Opin. Struct. Biol. 56, 146–154 (2019).
Ruhaak, L. R., Xu, G., Li, Q., Goonatilleke, E. & Lebrilla, C. B. Mass spectrometry approaches to glycomic and glycoproteomic analyses. Chem. Rev. 118, 7886–7930 (2018).
Narimatsu, H. et al. Current technologies for complex glycoproteomics and their applications to biology/disease-driven glycoproteomics. J. Proteome Res. 17, 4097–4112 (2018).
Yu, A. et al. Advances in mass spectrometry-based glycoproteomics. Electrophoresis 39, 3104–3122 (2018).
Cipollo, J. F. & Parsons, L. M. Glycomics and glycoproteomics of viruses: mass spectrometry applications and insights toward structure-function relationships. Mass Spectrom. Rev. 39, 371–409 (2020).
Suttapitugsakul, S., Sun, F. & Wu, R. Recent advances in glycoproteomic analysis by mass spectrometry. Anal. Chem. 92, 267–291 (2020).
Oliveira, T., Thaysen-Andersen, M., Packer, N. H. & Kolarich, D. The Hitchhiker’s guide to glycoproteomics. Biochem. Soc. Trans. 49, 1643–1662 (2021).
Ye, Z. & Vakhrushev, S. Y. The role of data-independent acquisition for glycoproteomics. Mol. Cell Proteom. 20, 100042 (2021).
Rangel-Angarita, V. & Malaker, S. A. Mucinomics as the next frontier of mass spectrometry. ACS Chem. Biol. 16, 1866–1883 (2021).
Parker, B. L. et al. Terminal galactosylation and sialylation switching on membrane glycoproteins upon TNF-alpha-induced insulin resistance in adipocytes. Mol. Cell Proteom. 15, 141–153 (2016). This is among the first studies to use glycomic, glycopeptide and PNGase F-treated glycopeptide information together to define glycan structural changes (site occupancy and glycan remodelling) at site-specific resolution.
King, S. L. et al. Characterizing the O-glycosylation landscape of human plasma, platelets, and endothelial cells. Blood Adv. 1, 429–442 (2017).
Liu, M. Q. et al. pGlyco 2.0 enables precision N-glycoproteomics with comprehensive quality control and one-step mass spectrometry for intact glycopeptide identification. Nat. Commun. 8, 438 (2017).
Yang, H., Yang, C. & Sun, T. Characterization of glycopeptides using a stepped higher-energy C-trap dissociation approach on a hybrid quadrupole orbitrap. Rapid Commun. Mass Spectrom. 32, 1353–1362 (2018).
Yang, S. et al. Deciphering protein O-glycosylation: solid-phase chemoenzymatic cleavage and enrichment. Anal. Chem. 90, 8261–8269 (2018).
Riley, N. M., Hebert, A. S., Westphall, M. S. & Coon, J. J. Capturing site-specific heterogeneity with large-scale N-glycoproteome analysis. Nat. Commun. 10, 1311 (2019).
Woo, C. M. et al. Mapping and quantification of over 2000 O-linked glycopeptides in activated human T cells with isotope-targeted glycoproteomics (Isotag). Mol. Cell Proteom. 17, 764–775 (2018).
Zhang, Y. et al. Systems analysis of singly and multiply O-glycosylated peptides in the human serum glycoproteome via EThcD and HCD mass spectrometry. J. Proteom. 170, 14–27 (2018).
Xiao, K. & Tian, Z. GPSeeker enables quantitative structural N-glycoproteomics for site- and structure-specific characterization of differentially expressed N-glycosylation in hepatocellular carcinoma. J. Proteome Res. 18, 2885–2895 (2019).
Darula, Z. & Medzihradszky, K. F. Carbamidomethylation side reactions may lead to glycan misassignments in glycopeptide analysis. Anal. Chem. 87, 6297–6302 (2015). A seminal study showing the importance of careful analysis of glycopeptide assignments and how suboptimal sample preparation conditions can compromise glycopeptide data sets.
Lee, L. Y. et al. Toward automated N-glycopeptide identification in glycoproteomics. J. Proteome Res. 15, 3904–3915 (2016).
Riley, N. M., Malaker, S. A. & Bertozzi, C. R. Electron-based dissociation is needed for O-glycopeptides derived from OpeRATOR proteolysis. Anal. Chem. 92, 14878–14884 (2020).
Zielinska, D. F., Gnad, F., Schropp, K., Wisniewski, J. R. & Mann, M. Mapping N-glycosylation sites across seven evolutionarily distant species reveals a divergent substrate proteome despite a common core machinery. Mol. Cell 46, 542–548 (2012).
Neubert, P. et al. Mapping the O-mannose glycoproteome in Saccharomyces cerevisiae. Mol. Cell. Proteom. 15, 1323–1337 (2016).
Bagdonaite, I. et al. Global mapping of O-glycosylation of Varicella zoster virus, human cytomegalovirus, and Epstein-Barr virus. J. Biol. Chem. 291, 12014–12028 (2016).
Madsen, T. D. et al. An atlas of O-linked glycosylation on peptide hormones reveals diverse biological roles. Nat. Commun. 11, 4033 (2020). Seminal work that describes the discovery of widespread O-glycosylation of endogenous peptide hormones with diverse biological functions.
Khetarpal, S. A. et al. Loss of Function of GALNT2 lowers high-density lipoproteins in humans, nonhuman primates, and rodents. Cell Metab. 24, 234–245 (2016).
Zhao, P. et al. Virus-receptor interactions of glycosylated SARS-CoV-2 spike and human ACE2 receptor. Cell Host Microbe 28, 586–601 e586 (2020).
Watanabe, Y., Allen, J. D., Wrapp, D., McLellan, J. S. & Crispin, M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science 369, 330–333 (2020).
Stavenhagen, K. et al. Site-specific N- and O-glycosylation analysis of atacicept. MAbs 11, 1053–1063 (2019).
Nason, R. et al. Display of the human mucinome with defined O-glycans by gene engineered cells. Nat. Commun. 12, 4070 (2021).
Yao, H. et al. Molecular architecture of the SARS-CoV-2 virus. Cell 183, 730–738.e13 (2020).
Shajahan, A., Supekar, N. T., Gleinich, A. S. & Azadi, P. Deducing the N- and O-glycosylation profile of the spike protein of novel coronavirus SARS-CoV-2. Glycobiology 30, 981–988 (2020).
Thaysen-Andersen, M. & Packer, N. H. Site-specific glycoproteomics confirms that protein structure dictates formation of N-glycan type, core fucosylation and branching. Glycobiology 22, 1440–1452 (2012).
Steentoft, C. et al. Mining the O-glycoproteome using zinc-finger nuclease-glycoengineered SimpleCell lines. Nat. Methods 8, 977–982 (2011). One of the first examples of a robust, high-throughput proteomic enrichment approach for the study of mucin O-glycosylation.
Steentoft, C. et al. Precision mapping of the human O-GalNAc glycoproteome through SimpleCell technology. EMBO J. 32, 1478–1488 (2013).
Bagdonaite, I. et al. A strategy for O-glycoproteomics of enveloped viruses–the O-glycoproteome of herpes simplex virus type 1. PLoS Pathog. 11, e1004784 (2015).
Schjoldager, K. T. et al. Deconstruction of O-glycosylation–GalNAc-T isoforms direct distinct subsets of the O-glycoproteome. EMBO Rep. 16, 1713–1722 (2015).
Wang, S. et al. Site-specific O-glycosylation of members of the low-density lipoprotein receptor superfamily enhances ligand interactions. J. Biol. Chem. 293, 7408–7422 (2018).
Narimatsu, Y. et al. Exploring regulation of protein O-glycosylation in isogenic human HEK293 cells by differential O-glycoproteomics. Mol. Cell Proteom. 18, 1396–1409 (2019).
Lavrsen, K. et al. De novo expression of human polypeptide N-acetylgalactosaminyltransferase 6 (GalNAc-T6) in colon adenocarcinoma inhibits the differentiation of colonic epithelium. J. Biol. Chem. 293, 1298–1314 (2018).
Bagdonaite, I. et al. O-glycan initiation directs distinct biological pathways and controls epithelial differentiation. EMBO Rep. 21, e48885 (2020).
Hintze, J. et al. Probing the contribution of individual polypeptide GalNAc-transferase isoforms to the O-glycoproteome by inducible expression in isogenic cell lines. J. Biol. Chem. 293, 19064–19077 (2018).
Ashraf Kharaz, Y. et al. Comparison between chaotropic and detergent-based sample preparation workflow in tendon for mass spectrometry analysis. Proteomics 17, 1700018 (2017).
Li, J. et al. Proteomics and N-glycoproteomics analysis of an extracellular matrix-based scaffold-human treated dentin matrix. J. Tissue Eng. Regen. Med. 13, 1164–1177 (2019).
Liu, Y. et al. Investigation of cell wall proteins of C. sinensis leaves by combining cell wall proteomics and N-glycoproteomics. BMC Plant. Biol. 21, 384 (2021).
Fang, P. et al. In-depth mapping of the mouse brain N-glycoproteome reveals widespread N-glycosylation of diverse brain proteins. Oncotarget 7, 38796–38809 (2016).
Arike, L., Holmen-Larsson, J. & Hansson, G. C. Intestinal Muc2 mucin O-glycosylation is affected by microbiota and regulated by differential expression of glycosyltranferases. Glycobiology 27, 318–328 (2017).
Schulz, B. L. et al. Glycosylation of sputum mucins is altered in cystic fibrosis patients. Glycobiology 17, 698–712 (2007).
Loo, R. R., Dales, N. & Andrews, P. C. The effect of detergents on proteins analyzed by electrospray ionization. Methods Mol. Biol. 61, 141–160 (1996).
Yeung, Y. G., Nieves, E., Angeletti, R. H. & Stanley, E. R. Removal of detergents from protein digests for mass spectrometry analysis. Anal. Biochem. 382, 135–137 (2008).
Woo, C. M., Iavarone, A. T., Spiciarich, D. R., Palaniappan, K. K. & Bertozzi, C. R. Isotope-targeted glycoproteomics (IsoTaG): a mass-independent platform for intact N- and O-glycopeptide discovery and analysis. Nat. Methods 12, 561–567 (2015).
Vester-Christensen, M. B. et al. Mining the O-mannose glycoproteome reveals cadherins as major O-mannosylated glycoproteins. Proc. Natl Acad. Sci. USA 110, 21018–21023 (2013).
Liu, J. et al. High-sensitivity N-glycoproteomic analysis of mouse brain tissue by protein extraction with a mild detergent of N-dodecyl beta-d-maltoside. Anal. Chem. 87, 2054–2057 (2015).
Malaker, S. A. et al. The mucin-selective protease StcE enables molecular and functional analysis of human cancer-associated mucins. Proc. Natl Acad. Sci. USA 116, 7278–7287 (2019). Seminal paper that introduces the concept of using mucin-selective proteases to access previously inaccessible regions of the glycoproteome.
Wu, F., Sun, D., Wang, N., Gong, Y. & Li, L. Comparison of surfactant-assisted shotgun methods using acid-labile surfactants and sodium dodecyl sulfate for membrane proteome analysis. Anal. Chim. Acta 698, 36–43 (2011).
Chang, Y. H. et al. New mass-spectrometry-compatible degradable surfactant for tissue proteomics. J. Proteome Res. 14, 1587–1599 (2015).
Glatter, T., Ahrne, E. & Schmidt, A. Comparison of different sample preparation protocols reveals lysis buffer-specific extraction biases in Gram-negative bacteria and human cells. J. Proteome Res. 14, 4472–4485 (2015).
Meissner, F., Scheltema, R. A., Mollenkopf, H. J. & Mann, M. Direct proteomic quantification of the secretome of activated immune cells. Science 340, 475–478 (2013).
Geiszler, D. J. et al. PTM-Shepherd: analysis and summarization of post-translational and chemical modifications from open search results. Mol. Cell Proteom. 20, 100018 (2020).
Wisniewski, J. R., Zougman, A., Nagaraj, N. & Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 (2009).
Zougman, A., Selby, P. J. & Banks, R. E. Suspension trapping (STrap) sample preparation method for bottom-up proteomic analysis. Proteomics 14, 1006-0 (2014).
HaileMariam, M. et al. S-Trap, an ultrafast sample-preparation approach for shotgun proteomics. J. Proteome Res. 17, 2917–2924 (2018).
Cao, J. et al. Multiplexed CuAAC Suzuki-Miyaura labeling for tandem activity-based chemoproteomic profiling. Anal. Chem. 93, 2610–2618 (2021).
Yan, T. et al. SP3-FAIMS chemoproteomics for high-coverage profiling of the human cysteinome. Chembiochem 22, 1841–1851 (2021).
Batth, T. S. et al. Protein aggregation capture on microparticles enables multipurpose proteomics sample preparation. Mol. Cell Proteom. 18, 1027–1035 (2019).
Leutert, M., Rodriguez-Mias, R. A., Fukuda, N. K. & Villen, J. R2-P2 rapid-robotic phosphoproteomics enables multidimensional cell signaling studies. Mol. Syst. Biol. 15, e9021 (2019).
Hughes, C. S. et al. Ultrasensitive proteome analysis using paramagnetic bead technology. Mol. Syst. Biol. 10, 757 (2014).
Zielinska, D. F., Gnad, F., Wisniewski, J. R. & Mann, M. Precision mapping of an in vivo N-glycoproteome reveals rigid topological and sequence constraints. Cell 141, 897–907 (2010).
Hughes, C. S. et al. Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nat. Protoc. 14, 68–85 (2019).
Fang, P. et al. A streamlined pipeline for multiplexed quantitative site-specific N-glycoproteomics. Nat. Commun. 11, 5268 (2020).
Kearney, C. J. et al. SUGAR-seq enables simultaneous detection of glycans, epitopes, and the transcriptome in single cells. Sci. Adv. 7, eabe3610 (2021).
Burt, R. A. et al. Novel antibodies for the simple and efficient enrichment of native O-GlcNAc modified peptides. Mol. Cell. Proteomics 20, 100167 (2021).
Radhakrishnan, P. et al. Immature truncated O-glycophenotype of cancer directly induces oncogenic features. Proc. Natl Acad. Sci. USA 111, E4066–E4075 (2014).
Dabelsteen, S. et al. Essential functions of glycans in human epithelia dissected by a CRISPR-Cas9-engineered human organotypic skin model. Dev. Cell 54, 669–684.e667 (2020).
van der Post, S., Thomsson, K. A. & Hansson, G. C. Multiple enzyme approach for the characterization of glycan modifications on the C-terminus of the intestinal MUC2mucin. J. Proteome Res. 13, 6013–6023 (2014).
Dodds, E. D., Seipert, R. R., Clowers, B. H., German, J. B. & Lebrilla, C. B. Analytical performance of immobilized pronase for glycopeptide footprinting and implications for surpassing reductionist glycoproteomics. J. Proteome Res. 8, 502–512 (2009).
Hoffmann, M., Marx, K., Reichl, U., Wuhrer, M. & Rapp, E. Site-specific O-glycosylation analysis of human blood plasma proteins. Mol. Cell. Proteom. 15, 624–641 (2016).
Larsen, M. R., Hojrup, P. & Roepstorff, P. Characterization of gel-separated glycoproteins using two-step proteolytic digestion combined with sequential microcolumns and mass spectrometry. Mol. Cell. Proteom. 4, 107–119 (2005).
Stavenhagen, K., Plomp, R. & Wuhrer, M. Site-specific protein N- and O-glycosylation analysis by a C18-porous graphitized carbon-liquid chromatography-electrospray ionization mass spectrometry approach using pronase treated glycopeptides. Anal. Chem. 87, 11691–11699 (2015).
Stavenhagen, K. et al. N- and O-glycosylation analysis of human C1-inhibitor reveals extensive mucin-type O-glycosylation. Mol. Cell. Proteom. 17, 1225–1238 (2018).
Shon, D. J., Kuo, A., Ferracane, M. J. & Malaker, S. A. Classification, structural biology, and applications of mucin domain-targeting proteases. Biochem. J. 478, 1585–1603 (2021).
Yang, W., Ao, M., Hu, Y., Li, Q. K. & Zhang, H. Mapping the O-glycoproteome using site-specific extraction of O-linked glycopeptides (EXoO). Mol. Syst. Biol. 14, e8486 (2018).
Shon, D. J. et al. An enzymatic toolkit for selective proteolysis, detection, and visualization of mucin-domain glycoproteins. Proc. Natl Acad. Sci. USA 117, 21299–21307 (2020).
Haurat, M. F. et al. The glycoprotease CpaA secreted by medically relevant acinetobacter species targets multiple O-linked host glycoproteins. mBio 11, e02033-20 (2020).
Vainauskas, S. et al. A broad-specificity O-glycoprotease that enables improved analysis of glycoproteins and glycopeptides containing intact complex O-glycans. Anal. Chem. 94, 1060–1069 (2022).
Kobata, A. Exo- and endoglycosidases revisited. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 89, 97–117 (2013).
Riley, N. M., Bertozzi, C. R. & Pitteri, S. J. A pragmatic guide to enrichment strategies for mass spectrometry-based glycoproteomics. Mol. Cell. Proteom. 20, 100029 (2020).
Hagglund, P., Bunkenborg, J., Elortza, F., Jensen, O. N. & Roepstorff, P. A new strategy for identification of N-glycosylated proteins and unambiguous assignment of their glycosylation sites using HILIC enrichment and partial deglycosylation. J. Proteome Res. 3, 556–566 (2004). Seminal work that introduces the use of HILIC for the enrichment and identification of glycopeptides from complex samples.
Stavenhagen, K. et al. Quantitative mapping of glycoprotein micro-heterogeneity and macro-heterogeneity: an evaluation of mass spectrometry signal strengths using synthetic peptides and glycopeptides. J. Mass Spectrom. 48, 627–639 (2013). A critical study providing some of the only experimental evidence on the impact of glycosylation events on the observed MS signal of glycopeptides compared with matching unglycosylated peptides at equal concentrations.
Gutierrez-Reyes, C. D. et al. Advances in mass spectrometry-based glycoproteomics: an update covering the period 2017–2021. Electrophoresis 43, 370–387 (2022).
Nilsson, J. et al. Enrichment of glycopeptides for glycan structure and attachment site identification. Nat. Methods 6, 809–811 (2009).
Van Lenten, L. & Ashwell, G. Studies on the chemical and enzymatic modification of glycoproteins. A general method for the tritiation of sialic acid-containing glycoproteins. J. Biol. Chem. 246, 1889–1894 (1971).
Zhang, H., Li, X. J., Martin, D. B. & Aebersold, R. Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat. Biotechnol. 21, 660–666 (2003). Seminal work that popularized the use of hydrazide-based enrichment coupled to deglycosylation to analyse N-linked glycosylation events.
Rafelson, M. E. Jr, Clauser, H. & Legault-Demare, J. Removal of sialic acid from serum gonadotropin by acidic and enzymic hydrolysis. Biochim. Biophys. Acta 47, 406–407 (1961).
Cooper, J. A., Smith, W., Bacila, M. & Medina, H. Galactose oxidase from Polyporus circinatus, Fr. J. Biol. Chem. 234, 445–448 (1959).
Gahmberg, C. G. & Hakomori, S. I. External labeling of cell surface galactose and galactosamine in glycolipid and glycoprotein of human erythrocytes. J. Biol. Chem. 248, 4311–4317 (1973).
Zeng, Y., Ramya, T. N., Dirksen, A., Dawson, P. E. & Paulson, J. C. High-efficiency labeling of sialylated glycoproteins on living cells. Nat. Methods 6, 207–209 (2009).
Shimaoka, H. et al. One-pot solid-phase glycoblotting and probing by transoximization for high-throughput glycomics and glycoproteomics. Chemistry 13, 1664–1673 (2007).
Ramya, T. N., Weerapana, E., Cravatt, B. F. & Paulson, J. C. Glycoproteomics enabled by tagging sialic acid- or galactose-terminated glycans. Glycobiology 23, 211–221 (2013).
Xiao, H., Chen, W., Smeekens, J. M. & Wu, R. An enrichment method based on synergistic and reversible covalent interactions for large-scale analysis of glycoproteins. Nat. Commun. 9, 1692 (2018).
Larsen, M. R., Jensen, S. S., Jakobsen, L. A. & Heegaard, N. H. Exploring the sialiome using titanium dioxide chromatography and mass spectrometry. Mol. Cell Proteom. 6, 1778–1787 (2007).
Palmisano, G. et al. Selective enrichment of sialic acid-containing glycopeptides using titanium dioxide chromatography with analysis by HILIC and mass spectrometry. Nat. Protoc. 5, 1974–1982 (2010).
Palmisano, G. et al. A novel method for the simultaneous enrichment, identification, and quantification of phosphopeptides and sialylated glycopeptides applied to a temporal profile of mouse brain development. Mol. Cell Proteom. 11, 1191–1202 (2012).
Zhang, H. et al. Simultaneous characterization of glyco- and phosphoproteomes of mouse brain membrane proteome with electrostatic repulsion hydrophilic interaction chromatography. Mol. Cell. Proteom. 9, 635–647 (2010).
Jensen, P. H., Mysling, S., Hojrup, P. & Jensen, O. N. Glycopeptide enrichment for MALDI-TOF mass spectrometry analysis by hydrophilic interaction liquid chromatography solid phase extraction (HILIC SPE). Methods Mol. Biol. 951, 131–144 (2013).
Neue, K., Mormann, M., Peter-Katalinic, J. & Pohlentz, G. Elucidation of glycoprotein structures by unspecific proteolysis and direct nanoESI mass spectrometric analysis of ZIC-HILIC-enriched glycopeptides. J. Proteome Res. 10, 2248–2260 (2011).
Selman, M. H., Hemayatkar, M., Deelder, A. M. & Wuhrer, M. Cotton HILIC SPE microtips for microscale purification and enrichment of glycans and glycopeptides. Anal. Chem. 83, 2492–2499 (2011).
Mysling, S., Palmisano, G., Hojrup, P. & Thaysen-Andersen, M. Utilizing ion-pairing hydrophilic interaction chromatography solid phase extraction for efficient glycopeptide enrichment in glycoproteomics. Anal. Chem. 82, 5598–5609 (2010). Critical study that investigates the impact of ion pairing on HILIC enrichment, which established the widely used gold standard protocol for HILIC enrichment.
Chalkley, R. J., Thalhammer, A., Schoepfer, R. & Burlingame, A. L. Identification of protein O-GlcNAcylation sites using electron transfer dissociation mass spectrometry on native peptides. Proc. Natl Acad. Sci. USA 106, 8894–8899 (2009).
Vosseller, K. et al. O-linked N-acetylglucosamine proteomics of postsynaptic density preparations using lectin weak affinity chromatography and mass spectrometry. Mol. Cell. Proteom. 5, 923–934 (2006).
Zachara, N. et al. in Essentials of Glycobiology Ch.19 (ed. Varki, A. et al.) (Cold Spring Harbor Laboratory Press, 2015).
Yurewicz, E. C., Pack, B. A. & Sacco, A. G. Porcine oocyte zona pellucida Mr 55,000 glycoproteins: identification of O-glycosylated domains. Mol. Reprod. Dev. 33, 182–188 (1992).
Steentoft, C., Bennett, E. P. & Clausen, H. Glycoengineering of human cell lines using zinc finger nuclease gene targeting: SimpleCells with homogeneous GalNAc O-glycosylation allow isolation of the O-glycoproteome by one-step lectin affinity chromatography. Methods Mol. Biol. 1022, 387–402 (2013).
Lee, A. et al. Combined antibody/lectin enrichment identifies extensive changes in the O-GlcNAc sub-proteome upon oxidative stress. J. Proteome Res. 15, 4318–4336 (2016).
Larsen, I. S. B. et al. Mammalian O-mannosylation of cadherins and plexins is independent of protein O-mannosyltransferases 1 and 2. J. Biol. Chem. 292, 11586–11598 (2017).
Larsen, I. S. B. et al. Discovery of an O-mannosylation pathway selectively serving cadherins and protocadherins. Proc. Natl Acad. Sci. USA 114, 11163–11168 (2017).
Pedowitz, N. J. & Pratt, M. R. Design and synthesis of metabolic chemical reporters for the visualization and identification of glycoproteins. RSC Chem. Biol. 2, 306–321 (2021).
Critcher, M., O’Leary, T. & Huang, M. L. Glycoengineering: scratching the surface. Biochem. J. 478, 703–719 (2021).
Sletten, E. M. & Bertozzi, C. R. Bioorthogonal chemistry: fishing for selectivity in a sea of functionality. Angew. Chem. Int. Ed. 48, 6974–6998 (2009).
Parker, C. G. & Pratt, M. R. Click chemistry in proteomic investigations. Cell 180, 605–632 (2020).
Hang, H. C., Yu, C., Kato, D. L. & Bertozzi, C. R. A metabolic labeling approach toward proteomic analysis of mucin-type O-linked glycosylation. Proc. Natl Acad. Sci. USA 100, 14846–14851 (2003).
Prescher, J. A. & Bertozzi, C. R. Chemistry in living systems. Nat. Chem. Biol. 1, 13–21 (2005).
Hang, H. C., Yu, C., Pratt, M. R. & Bertozzi, C. R. Probing glycosyltransferase activities with the Staudinger ligation. J. Am. Chem. Soc. 126, 6–7 (2004).
Boyce, M. et al. Metabolic cross-talk allows labeling of O-linked beta-N-acetylglucosamine-modified proteins via the N-acetylgalactosamine salvage pathway. Proc. Natl Acad. Sci. USA 108, 3141–3146 (2011).
Alfaro, J. F. et al. Tandem mass spectrometry identifies many mouse brain O-GlcNAcylated proteins including EGF domain-specific O-GlcNAc transferase targets. Proc. Natl Acad. Sci. USA 109, 7280–7285 (2012).
Cioce, A., Malaker, S. A. & Schumann, B. Generating orthogonal glycosyltransferase and nucleotide sugar pairs as next-generation glycobiology tools. Curr. Opin. Chem. Biol. 60, 66–78 (2020).
Choi, J. et al. Engineering orthogonal polypeptide GalNAc-transferase and UDP-sugar pairs. J. Am. Chem. Soc. 141, 13442–13453 (2019).
Schumann, B. et al. Bump-and-hole engineering identifies specific substrates of glycosyltransferases in living cells. Mol. Cell 78, 824–834.e15 (2020). One of the first demonstrations of bump-and-hole engineering for the identification of mucin O-glycosylation events in living cells.
Debets, M. F. et al. Metabolic precision labeling enables selective probing of O-linked N-acetylgalactosamine glycosylation. Proc. Natl Acad. Sci. USA 117, 25293–25301 (2020).
Calle, B. et al. Benefits of chemical sugar modifications introduced by click chemistry for glycoproteomic analyses. J. Am. Soc. Mass Spectrom. 32, 2366–2375 (2021).
Shishkova, E., Hebert, A. S. & Coon, J. J. Now, more than ever, proteomics needs better chromatography. Cell Syst. 3, 321–324 (2016).
Ozohanics, O., Turiak, L., Puerta, A., Vekey, K. & Drahos, L. High-performance liquid chromatography coupled to mass spectrometry methodology for analyzing site-specific N-glycosylation patterns. J. Chromatogr. A 1259, 200–212 (2012).
Wang, B., Tsybovsky, Y., Palczewski, K. & Chance, M. R. Reliable determination of site-specific in vivo protein N-glycosylation based on collision-induced MS/MS and chromatographic retention time. J. Am. Soc. Mass Spectrom. 25, 729–741 (2014).
Kozlik, P., Goldman, R. & Sanda, M. Study of structure-dependent chromatographic behavior of glycopeptides using reversed phase nanoLC. Electrophoresis 38, 2193–2199 (2017).
Klein, J. & Zaia, J. Relative retention time estimation improves N-glycopeptide identifications by LC-MS/MS. J. Proteome Res. 19, 2113–2121 (2020).
Ang, E., Neustaeter, H., Spicer, V., Perreault, H. & Krokhin, O. Retention time prediction for glycopeptides in reversed-phase chromatography for glycoproteomic applications. Anal. Chem. 91, 13360–13366 (2019).
Choo, M. S., Wan, C., Rudd, P. M. & Nguyen-Khuong, T. GlycopeptideGraphMS: improved glycopeptide detection and identification by exploiting graph theoretical patterns in mass and retention time. Anal. Chem. 91, 7236–7244 (2019).
Gutierrez Reyes, C. D., Jiang, P., Donohoo, K., Atashi, M. & Mechref, Y. S. Glycomics and glycoproteomics: approaches to address isomeric separation of glycans and glycopeptides. J. Sep. Sci. 44, 403–425 (2021).
Molnarova, K., Duris, A., Jecmen, T. & Kozlik, P. Comparison of human IgG glycopeptides separation using mixed-mode hydrophilic interaction/ion-exchange liquid chromatography and reversed-phase mode. Anal. Bioanal. Chem. 413, 4321–4328 (2021).
Wohlgemuth, J., Karas, M., Jiang, W., Hendriks, R. & Andrecht, S. Enhanced glyco-profiling by specific glycopeptide enrichment and complementary monolithic nano-LC (ZIC-HILIC/RP18e)/ESI-MS analysis. J. Sep. Sci. 33, 880–890 (2010).
Ji, E. S. et al. Isomer separation of sialylated O- and N-linked glycopeptides using reversed-phase LC-MS/MS at high temperature. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 1110–1111, 101–107 (2019).
Zauner, G., Koeleman, C. A., Deelder, A. M. & Wuhrer, M. Protein glycosylation analysis by HILIC-LC-MS of Proteinase K-generated N- and O-glycopeptides. J. Sep. Sci. 33, 903–910 (2010).
Sun, N., Wu, H., Chen, H., Shen, X. & Deng, C. Advances in hydrophilic nanomaterials for glycoproteomics. Chem. Commun. 55, 10359–10375 (2019).
van der Burgt, Y. E. M., Siliakus, K. M., Cobbaert, C. M. & Ruhaak, L. R. HILIC-MRM-MS for linkage-specific separation of sialylated glycopeptides to quantify prostate-specific antigen proteoforms. J. Proteome Res. 19, 2708–2716 (2020).
Molnarova, K. & Kozlik, P. Comparison of different HILIC stationary phases in the separation of hemopexin and immunoglobulin G glycopeptides and their isomers. Molecules 25, 4655 (2020).
Bapiro, T. E., Richards, F. M. & Jodrell, D. I. Understanding the complexity of porous graphitic carbon (PGC) chromatography: modulation of mobile-stationary phase interactions overcomes loss of retention and reduces variability. Anal. Chem. 88, 6190–6194 (2016).
Hinneburg, H. et al. Post-column make-up flow (PCMF) enhances the performance of capillary-flow PGC-LC-MS/MS-based glycomics. Anal. Chem. 91, 4559–4567 (2019).
Alley, W. R. Jr, Mechref, Y. & Novotny, M. V. Use of activated graphitized carbon chips for liquid chromatography/mass spectrometric and tandem mass spectrometric analysis of tryptic glycopeptides. Rapid Commun. Mass Spectrom. 23, 495–505 (2009).
West, C., Elfakir, C. & Lafosse, M. Porous graphitic carbon: a versatile stationary phase for liquid chromatography. J. Chromatogr. A 1217, 3201–3216 (2010).
Xue, Y. et al. Study on behaviors and performances of universal N-glycopeptide enrichment methods. Analyst 143, 1870–1880 (2018).
Froehlich, J. W. et al. Nano-LC-MS/MS of glycopeptides produced by nonspecific proteolysis enables rapid and extensive site-specific glycosylation determination. Anal. Chem. 83, 5541–5547 (2011).
Nwosu, C. C. et al. In-gel nonspecific proteolysis for elucidating glycoproteins: a method for targeted protein-specific glycosylation analysis in complex protein mixtures. Anal. Chem. 85, 956–963 (2013).
Huang, J. et al. Site-specific glycosylation of secretory immunoglobulin A from human colostrum. J. Proteome Res. 14, 1335–1349 (2015).
Lee, J. et al. Designation of fingerprint glycopeptides for targeted glycoproteomic analysis of serum haptoglobin: insights into gastric cancer biomarker discovery. Anal. Bioanal. Chem. 410, 1617–1629 (2018).
Hua, S. et al. Glyco-analytical multispecific proteolysis (Glyco-AMP): a simple method for detailed and quantitative glycoproteomic characterization. J. Proteome Res. 12, 4414–4423 (2013).
Chen, R., Stupak, J., Williamson, S., Twine, S. M. & Li, J. Online porous graphic carbon chromatography coupled with tandem mass spectrometry for post-translational modification analysis. Rapid Commun. Mass Spectrom. 33, 1240–1247 (2019).
Zhu, R., Huang, Y., Zhao, J., Zhong, J. & Mechref, Y. Isomeric separation of N-glycopeptides derived from glycoproteins by porous graphitic carbon (PGC) LC-MS/MS. Anal. Chem. 92, 9556–9565 (2020).
Kammeijer, G. S. et al. Dopant enriched nitrogen gas combined with sheathless capillary electrophoresis-electrospray ionization-mass spectrometry for improved sensitivity and repeatability in glycopeptide analysis. Anal. Chem. 88, 5849–5856 (2016).
Qu, Y. et al. Sensitive and fast characterization of site-specific protein glycosylation with capillary electrophoresis coupled to mass spectrometry. Talanta 179, 22–27 (2018).
Pont, L. et al. Site-specific N-linked glycosylation analysis of human carcinoembryonic antigen by sheathless capillary electrophoresis-tandem mass spectrometry. J. Proteome Res. 20, 1666–1675 (2021).
Qu, Y., Sun, L., Zhang, Z. & Dovichi, N. J. Site-specific glycan heterogeneity characterization by hydrophilic interaction liquid chromatography solid-phase extraction, reversed-phase liquid chromatography fractionation, and capillary zone electrophoresis-electrospray ionization-tandem mass spectrometry. Anal. Chem. 90, 1223–1233 (2018).
Khatri, K. et al. Microfluidic capillary electrophoresis-mass spectrometry for analysis of monosaccharides, oligosaccharides, and glycopeptides. Anal. Chem. 89, 6645–6655 (2017).
Kammeijer, G. S. M. et al. Sialic acid linkage differentiation of glycopeptides using capillary electrophoresis-electrospray ionization-mass spectrometry. Sci. Rep. 7, 3733 (2017).
Melzer, T., Wimmer, B., Bock, S., Posch, T. N. & Huhn, C. Challenges and applications of isotachophoresis coupled to mass spectrometry: a review. Electrophoresis 41, 1045–1059 (2020).
Fenn, L. S. & McLean, J. A. Structural separations by ion mobility-MS for glycomics and glycoproteomics. Methods Mol. Biol. 951, 171–194 (2013).
Mookherjee, A. & Guttman, M. Bridging the structural gap of glycoproteomics with ion mobility spectrometry. Curr. Opin. Chem. Biol. 42, 86–92 (2018).
Chen, Z., Glover, M. S. & Li, L. Recent advances in ion mobility-mass spectrometry for improved structural characterization of glycans and glycoconjugates. Curr. Opin. Chem. Biol. 42, 1–8 (2018).
Struwe, W. B. & Harvey, D. J. Ion mobility-mass spectrometry of glycoconjugates. Methods Mol. Biol. 2084, 203–219 (2020).
Sarbu, M., Zhu, F., Peter-Katalinic, J., Clemmer, D. E. & Zamfir, A. D. Application of ion mobility tandem mass spectrometry to compositional and structural analysis of glycopeptides extracted from the urine of a patient diagnosed with Schindler disease. Rapid Commun. Mass Spectrom. 29, 1929–1937 (2015).
Kolli, V., Schumacher, K. N. & Dodds, E. D. Ion mobility-resolved collision-induced dissociation and electron transfer dissociation of N-glycopeptides: gathering orthogonal connectivity information from a single mass-selected precursor ion population. Analyst 142, 4691–4702 (2017).
Gelb, A. S., Lai, R., Li, H. & Dodds, E. D. Composition and charge state influence on the ion-neutral collision cross sections of protonated N-linked glycopeptides: an experimental and theoretical deconstruction of coulombic repulsion vs. charge solvation effects. Analyst 144, 5738–5747 (2019).
Barroso, A. et al. Evaluation of ion mobility for the separation of glycoconjugate isomers due to different types of sialic acid linkage, at the intact glycoprotein, glycopeptide and glycan level. J. Proteom. 173, 22–31 (2018).
Pallister, E. G. et al. Utility of ion-mobility spectrometry for deducing branching of multiply charged glycans and glycopeptides in a high-throughput positive ion LC-FLR-IMS-MS workflow. Anal. Chem. 92, 15323–15335 (2020).
Creese, A. J. & Cooper, H. J. Separation and identification of isomeric glycopeptides by high field asymmetric waveform ion mobility spectrometry. Anal. Chem. 84, 2597–2601 (2012).
Campbell, J. L. et al. Analyzing glycopeptide isomers by combining differential mobility spectrometry with electron- and collision-based tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 28, 1374–1381 (2017).
Pathak, P., Baird, M. A. & Shvartsburg, A. A. High-resolution ion mobility separations of isomeric glycoforms with variations on the peptide and glycan levels. J. Am. Soc. Mass Spectrom. 31, 1603–1609 (2020).
Wu, R. et al. Fine adjustment of gas modifier loadings for separation of epimeric glycopeptides using differential ion mobility spectrometry mass spectrometry. Rapid Commun. Mass Spectrom. 34, e8751 (2020).
Both, P. et al. Discrimination of epimeric glycans and glycopeptides using IM-MS and its potential for carbohydrate sequencing. Nat. Chem. 6, 65–74 (2014).
Hinneburg, H. et al. Distinguishing N-acetylneuraminic acid linkage isomers on glycopeptides by ion mobility-mass spectrometry. Chem. Commun. 52, 4381–4384 (2016).
Glaskin, R. S., Khatri, K., Wang, Q., Zaia, J. & Costello, C. E. Construction of a database of collision cross section values for glycopeptides, glycans, and peptides determined by IM-MS. Anal. Chem. 89, 4452–4460 (2017).
Feng, X. et al. Relative quantification of N-glycopeptide sialic acid linkage isomers by ion mobility mass spectrometry. Anal. Chem. 93, 15617–15625 (2021).
Ahmad Izaham, A. R. et al. What are we missing by using hydrophilic enrichment? improving bacterial glycoproteome coverage using total proteome and FAIMS analyses. J. Proteome Res. 20, 599–612 (2021).
Fang, P. et al. Evaluation and optimization of high-field asymmetric waveform ion-mobility spectrometry for multiplexed quantitative site-specific N-glycoproteomics. Anal. Chem. https://doi.org/10.1021/acs.analchem.1c00802 (2021).
Brown, C. J. et al. Glycoproteomic analysis of human urinary exosomes. Anal. Chem. 92, 14357–14365 (2020).
Lin, Y. et al. A panel of glycopeptides as candidate biomarkers for early diagnosis of NASH hepatocellular carcinoma using a stepped HCD method and PRM evaluation. J. Proteome Res. 20, 3278–3289 (2021).
Cho, K. C., Chen, L., Hu, Y., Schnaubelt, M. & Zhang, H. Developing workflow for simultaneous analyses of phosphopeptides and glycopeptides. ACS Chem. Biol. 14, 58–66 (2019).
Mao, J. et al. A new searching strategy for the identification of O-linked glycopeptides. Anal. Chem. 91, 3852–3859 (2019).
Shu, Q. et al. Large-scale identification of N-linked intact glycopeptides in human serum using HILIC enrichment and spectral library search. Mol. Cell. Proteom. 19, 672–689 (2020).
Alagesan, K., Hoffmann, M., Rapp, E. & Kolarich, D. Glycoproteomics technologies in glycobiotechnology. Adv. Biochem. Eng. Biotechnol. 175, 413–434 (2021).
Kawahara, R. et al. Distinct urinary glycoprotein signatures in prostate cancer patients. Oncotarget 9, 33077–33097 (2018).
Lu, J. et al. Determination of N-glycopeptides by hydrophilic interaction liquid chromatography and porous graphitized carbon chromatography with mass spectrometry detection. Anal. Lett. 50, 315–324 (2017).
Lewandrowski, U. & Sickmann, A. Online dual gradient reversed-phase/porous graphitized carbon nanoHPLC for proteomic applications. Anal. Chem. 82, 5391–5396 (2010).
Stavenhagen, K., Hinneburg, H., Kolarich, D. & Wuhrer, M. Site-specific N- and O-glycopeptide analysis using an integrated C18-PGC-LC-ESI-QTOF-MS/MS approach. Methods Mol. Biol. 1503, 109–119 (2017).
Zhao, Y. et al. Online two-dimensional porous graphitic carbon/reversed phase liquid chromatography platform applied to shotgun proteomics and glycoproteomics. Anal. Chem. 86, 12172–12179 (2014).
Aebersold, R. & Mann, M. Mass-spectrometric exploration of proteome structure and function. Nature 537, 347–355 (2016).
Xiao, H., Sun, F., Suttapitugsakul, S. & Wu, R. Global and site-specific analysis of protein glycosylation in complex biological systems with mass spectrometry. Mass. Spectrom. Rev. 38, 356–379 (2019).
Dodds, E. D. Gas-phase dissociation of glycosylated peptide ions. Mass. Spectrom. Rev. 31, 666–682 (2012).
Biemann, K. Appendix 5. Nomenclature for peptide fragment ions (positive ions). Methods Enzymol. 193, 886–887 (1990).
Reiding, K. R., Bondt, A., Franc, V. & Heck, A. J. R. The benefits of hybrid fragmentation methods for glycoproteomics. TrAC Trends Anal. Chem. 108, 260–268 (2018).
Domon, B. & Costello, C. E. A systematic nomenclature for carbohydrate fragmentations in FAB-MS/MS spectra of glycoconjugates. Glycoconj. J. 5, 397–409 (1988). Seminal work that outlines the glycan/glycoconjugate fragmentation nomenclature widely used across glycoproteomics studies.
Olsen, J. V. et al. Higher-energy C-trap dissociation for peptide modification analysis. Nat. Methods 4, 709–712 (2007).
Wells, J. M. & McLuckey, S. A. Collision-induced dissociation (CID) of peptides and proteins. Methods Enzymol. 402, 148–185 (2005).
Scott, N. E. et al. Simultaneous glycan-peptide characterization using hydrophilic interaction chromatography and parallel fragmentation by CID, higher energy collisional dissociation, and electron transfer dissociation MS applied to the N-linked glycoproteome of Campylobacter jejuni. Mol. Cell. Proteom. 10, M000031-MCP000201 (2011).
Cao, L. et al. Characterization of intact N- and O-linked glycopeptides using higher energy collisional dissociation. Anal. Biochem. 452, 96–102 (2014).
Hinneburg, H. et al. The art of destruction: optimizing collision energies in quadrupole-time of flight (Q-TOF) instruments for glycopeptide-based glycoproteomics. J. Am. Soc. Mass Spectrom. 27, 507–519 (2016).
Hoffmann, M. et al. The fine art of destruction: a guide to in-depth glycoproteomic analyses-exploiting the diagnostic potential of fragment ions. Proteomics 18, e1800282 (2018).
Kolli, V. & Dodds, E. D. Energy-resolved collision-induced dissociation pathways of model N-linked glycopeptides: implications for capturing glycan connectivity and peptide sequence in a single experiment. Analyst 139, 2144–2153 (2014).
Aboufazeli, F. & Dodds, E. D. Precursor ion survival energies of protonated N-glycopeptides and their weak dependencies on high mannose N-glycan composition in collision-induced dissociation. Analyst 143, 4459–4468 (2018).
Kelly, M. I. & Dodds, E. D. Parallel determination of polypeptide and oligosaccharide connectivities by energy-resolved collison-induced dissociation of protonated O-glycopeptides derived from nonspecific proteolysis. J. Am. Soc. Mass Spectrom. 31, 624–632 (2020).
Riley, N. M., Malaker, S. A., Driessen, M. & Bertozzi, C. R. Optimal dissociation methods differ for N- and O-glycopeptides. J. Proteome Res. 19, 3286–3301 (2020). A systematic study that outlines the benefits and trade-offs of using various fragmentation methods for the analysis of N-linked and O-linked glycopeptides.
Kolli, V., Roth, H. A., De La Cruz, G., Fernando, G. S. & Dodds, E. D. The role of proton mobility in determining the energy-resolved vibrational activation/dissociation channels of N-glycopeptide ions. Anal. Chim. Acta 896, 85–92 (2015).
You, X., Qin, H. & Ye, M. Recent advances in methods for the analysis of protein o-glycosylation at proteome level. J. Sep. Sci. 41, 248–261 (2018).
Acs, A., Ozohanics, O., Vekey, K., Drahos, L. & Turiak, L. Distinguishing core and antenna fucosylated glycopeptides based on low-energy tandem mass spectra. Anal. Chem. 90, 12776–12782 (2018).
Wang, Y. & Tian, Z. New energy setup strategy for intact N-glycopeptides characterization using higher-energy collisional dissociation. J. Am. Soc. Mass Spectrom. 31, 651–657 (2020).
Macias, L. A., Santos, I. C. & Brodbelt, J. S. Ion activation methods for peptides and proteins. Anal. Chem. 92, 227–251 (2020).
Thaysen-Andersen, M., Wilkinson, B. L., Payne, R. J. & Packer, N. H. Site-specific characterisation of densely O-glycosylated mucin-type peptides using electron transfer dissociation ESI-MS/MS. Electrophoresis 32, 3536–3545 (2011).
Darula, Z., Sherman, J. & Medzihradszky, K. F. How to dig deeper? Improved enrichment methods for mucin core-1 type glycopeptides. Mol. Cell. Proteom. 11, O111 016774 (2012).
Zhu, Z., Su, X., Clark, D. F., Go, E. P. & Desaire, H. Characterizing O-linked glycopeptides by electron transfer dissociation: fragmentation rules and applications in data analysis. Anal. Chem. 85, 8403–8411 (2013).
Mechref, Y. Use of CID/ETD mass spectrometry to analyze glycopeptides. Curr. Protoc. Protein Sci. https://doi.org/10.1002/0471140864.ps1211s68 (2012).
Riley, N. M. & Coon, J. J. The role of electron transfer dissociation in modern proteomics. Anal. Chem. 90, 40–64 (2018).
Alagesan, K., Hinneburg, H., Seeberger, P. H., Silva, D. V. & Kolarich, D. Glycan size and attachment site location affect electron transfer dissociation (ETD) fragmentation and automated glycopeptide identification. Glycoconj. J. 36, 487–493 (2019).
Yu, Q. et al. Electron-transfer/higher-energy collision dissociation (EThcD)-enabled intact glycopeptide/glycoproteome characterization. J. Am. Soc. Mass Spectrom. 28, 1751–1764 (2017).
Swaney, D. L. et al. Supplemental activation method for high-efficiency electron-transfer dissociation of doubly protonated peptide precursors. Anal. Chem. 79, 477–485 (2007).
Pap, A., Klement, E., Hunyadi-Gulyas, E., Darula, Z. & Medzihradszky, K. F. Status report on the high-throughput characterization of complex intact O-glycopeptide mixtures. J. Am. Soc. Mass Spectrom. 29, 1210–1220 (2018).
Zhang, L. & Reilly, J. P. Extracting both peptide sequence and glycan structural information by 157 nm photodissociation of N-linked glycopeptides. J. Proteome Res. 8, 734–742 (2009).
Madsen, J. A. et al. Concurrent automated sequencing of the glycan and peptide portions of O-linked glycopeptide anions by ultraviolet photodissociation mass spectrometry. Anal. Chem. 85, 9253–9261 (2013).
Halim, M. A. et al. Ultraviolet, infrared, and high-low energy photodissociation of post-translationally modified peptides. J. Am. Soc. Mass Spectrom. 29, 270–283 (2018).
Escobar, E. E. et al. Precision mapping of O-linked N-acetylglucosamine sites in proteins using ultraviolet photodissociation mass spectrometry. J. Am. Chem. Soc. 142, 11569–11577 (2020).
Dang, L. et al. Mapping human N-linked glycoproteins and glycosylation sites using mass spectrometry. Trends Anal. Chem. 114, 143–150 (2019).
Darula, Z., Pap, A. & Medzihradszky, K. F. Extended sialylated O-glycan repertoire of human urinary glycoproteins discovered and characterized using electron-transfer/higher-energy collision dissociation. J. Proteome Res. 18, 280–291 (2019).
Wu, S. W., Pu, T. H., Viner, R. & Khoo, K. H. Novel LC-MS(2) product dependent parallel data acquisition function and data analysis workflow for sequencing and identification of intact glycopeptides. Anal. Chem. 86, 5478–5486 (2014).
Singh, C., Zampronio, C. G., Creese, A. J. & Cooper, H. J. Higher energy collision dissociation (HCD) product ion-triggered electron transfer dissociation (ETD) mass spectrometry for the analysis of N-linked glycoproteins. J. Proteome Res. 11, 4517–4525 (2012).
Saba, J., Dutta, S., Hemenway, E. & Viner, R. Increasing the productivity of glycopeptides analysis by using higher-energy collision dissociation-accurate mass-product-dependent electron transfer dissociation. Int. J. Proteom. 2012, 560391 (2012). Critical work that outlines the first example of using oxonium fragment ions to trigger the collection of electron transfer dissociation data.
Zhou, C. & Schulz, B. L. Glycopeptide variable window SWATH for improved data independent acquisition glycoprotein analysis. Anal. Biochem. 597, 113667 (2020).
Chang, D., Klein, J. A., Nalehua, M. R., Hackett, W. E. & Zaia, J. Data-independent acquisition mass spectrometry for site-specific glycoproteomics characterization of SARS-CoV-2 spike protein. Anal. Bioanal. Chem. 413, 7305–7318 (2021).
Zacchi, L. F. & Schulz, B. L. SWATH-MS glycoproteomics reveals consequences of defects in the glycosylation machinery. Mol. Cell. Proteom. 15, 2435–2447 (2016).
Sanda, M. & Goldman, R. Data independent analysis of IgG glycoforms in samples of unfractionated human plasma. Anal. Chem. 88, 10118–10125 (2016).
Sanda, M., Zhang, L., Edwards, N. J. & Goldman, R. Site-specific analysis of changes in the glycosylation of proteins in liver cirrhosis using data-independent workflow with soft fragmentation. Anal. Bioanal. Chem. 409, 619–627 (2017).
Pan, K. T., Chen, C. C., Urlaub, H. & Khoo, K. H. Adapting data-independent acquisition for mass spectrometry-based protein site-specific N-glycosylation analysis. Anal. Chem. 89, 4532–4539 (2017).
Lin, C. H., Krisp, C., Packer, N. H. & Molloy, M. P. Development of a data independent acquisition mass spectrometry workflow to enable glycopeptide analysis without predefined glycan compositional knowledge. J. Proteom. 172, 68–75 (2018).
Pegg, C. L. et al. Quantitative data-independent acquisition glycoproteomics of sparkling wine. Mol. Cell. Proteom. 20, 100020 (2020).
Madsen, J. A., Farutin, V., Lin, Y. Y., Smith, S. & Capila, I. Data-independent oxonium ion profiling of multi-glycosylated biotherapeutics. MAbs 10, 968–978 (2018).
Dong, M. et al. Data-independent acquisition-based mass spectrometry (DIA-MS) for quantitative analysis of intact N-linked glycopeptides. Anal. Chem. 93, 13774–13782 (2021).
Ye, Z., Mao, Y., Clausen, H. & Vakhrushev, S. Y. Glyco-DIA: a method for quantitative O-glycoproteomics with in silico-boosted glycopeptide libraries. Nat. Methods 16, 902–910 (2019).
Baba, T. et al. Dissociation of biomolecules by an intense low-energy electron beam in a high sensitivity time-of-flight mass spectrometer. J. Am. Soc. Mass Spectrom. 32, 1964–1975 (2021).
Beckman, J. S. et al. Improved protein and PTM characterization with a practical electron-based fragmentation on Q-TOF instruments. J. Am. Soc. Mass Spectrom. 32, 2081–2091 (2021).
Reiding, K. R., Lin, Y. H., van Alphen, F. P. J., Meijer, A. B. & Heck, A. J. R. Neutrophil azurophilic granule glycoproteins are distinctively decorated by atypical pauci- and phosphomannose glycans. Commun. Biol. 4, 1012 (2021).
Ankney, J. A., Muneer, A. & Chen, X. Relative and absolute quantitation in mass spectrometry-based proteomics. Annu. Rev. Anal. Chem. 11, 49–77 (2018).
Ong, S. E. et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteom. 1, 376–386 (2002).
Deeb, S. J., Cox, J., Schmidt-Supprian, M. & Mann, M. N-linked glycosylation enrichment for in-depth cell surface proteomics of diffuse large B-cell lymphoma subtypes. Mol. Cell. Proteom. 13, 240–251 (2014).
Qin, W. et al. Quantitative time-resolved chemoproteomics reveals that stable O-GlcNAc regulates box C/D snoRNP biogenesis. Proc. Natl Acad. Sci. USA 114, E6749–E6758 (2017).
Hsu, J. L., Huang, S. Y., Chow, N. H. & Chen, S. H. Stable-isotope dimethyl labeling for quantitative proteomics. Anal. Chem. 75, 6843–6852 (2003).
Boersema, P. J., Aye, T. T., van Veen, T. A., Heck, A. J. & Mohammed, S. Triplex protein quantification based on stable isotope labeling by peptide dimethylation applied to cell and tissue lysates. Proteomics 8, 4624–4632 (2008).
Jung, J. et al. Deuterium-free, three-plexed peptide diethylation for highly accurate quantitative proteomics. J. Proteome Res. 18, 1078–1087 (2019).
Boersema, P. J., Raijmakers, R., Lemeer, S., Mohammed, S. & Heck, A. J. Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat. Protoc. 4, 484–494 (2009).
Thompson, A. et al. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 75, 1895–1904 (2003).
Franken, H. et al. Thermal proteome profiling for unbiased identification of direct and indirect drug targets using multiplexed quantitative mass spectrometry. Nat. Protoc. 10, 1567–1593 (2015).
Zhang, L. & Elias, J. E. Relative protein quantification using tandem mass tag mass spectrometry. Methods Mol. Biol. 1550, 185–198 (2017).
Ross, P. L. et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteom. 3, 1154–1169 (2004).
Ting, L., Rad, R., Gygi, S. P. & Haas, W. MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat. Methods 8, 937–940 (2011).
McAlister, G. C. et al. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem. 86, 7150–7158 (2014).
Li, J. et al. TMTpro-18plex: the expanded and complete set of TMTpro reagents for sample multiplexing. J. Proteome Res. 20, 2964–2972 (2021).
Viner, R. I., Snovida, S., Bodnar, E., Perreault, H. & Saba, J. A novel workflow for glycopeptide analysis using cellulose-based separation cartridges, TMT-labeling and LTQ orbitrap ETD. J. Biomol. Tech. 21, S25–S25 (2010).
Zecha, J. et al. TMT labeling for the masses: a robust and cost-efficient, in-solution labeling approach. Mol. Cell. Proteom. 18, 1468–1478 (2019).
Mao, Y. et al. Systematic evaluation of fragmentation methods for unlabeled and isobaric mass tag-labeled O-glycopeptides. Anal. Chem. 93, 11167–11175 (2021).
Wang, S. et al. Quantitative proteomics identifies altered O-GlcNAcylation of structural, synaptic and memory-associated proteins in Alzheimer’s disease. J. Pathol. 243, 78–88 (2017).
White, C. W. 3rd et al. Age-related loss of neural stem cell O-GlcNAc promotes a glial fate switch through STAT3 activation. Proc. Natl Acad. Sci. USA 117, 22214–22224 (2020).
Blazev, R. et al. Integrated glycoproteomics identifies a role of N-glycosylation and galectin-1 on myogenesis and muscle development. Mol. Cell. Proteom. 20, 100030 (2020).
Parker, B. L. et al. Multiplexed temporal quantification of the exercise-regulated plasma peptidome. Mol. Cell. Proteom. 16, 2055–2068 (2017).
Nilsson, J. et al. Synthetic standard aided quantification and structural characterization of amyloid-beta glycopeptides enriched from cerebrospinal fluid of Alzheimer’s disease patients. Sci. Rep. 9, 5522 (2019).
Kim, K. H. et al. Absolute quantification of N-glycosylation of alpha-fetoprotein using parallel reaction monitoring with stable isotope-labeled N-glycopeptide as an internal standard. Anal. Chem. 92, 12588–12595 (2020).
Eng, J. K., Searle, B. C., Clauser, K. R. & Tabb, D. L. A face in the crowd: recognizing peptides through database search. Mol. Cell. Proteom. 10, R111.009522 (2011).
Sadygov, R. G., Cociorva, D. & Yates, J. R. III Large-scale database searching using tandem mass spectra: looking up the answer in the back of the book. Nat. Methods 1, 195–202 (2004).
Perkins, D. N., Pappin, D. J., Creasy, D. M. & Cottrell, J. S. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 (1999).
Eng, J. K., McCormack, A. L. & Yates, J. R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989 (1994).
Cox, J. et al. Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 (2011).
Dorfer, V. et al. MS Amanda, a universal identification algorithm optimized for high accuracy tandem mass spectra. J. Proteome Res. 13, 3679–3684 (2014).
Stadlmann, J., Hoi, D. M., Taubenschmid, J., Mechtler, K. & Penninger, J. M. Analysis of PNGase F-resistant N-glycopeptides using sugarQb for Proteome Discoverer 2.1 reveals cryptic substrate specificities. Proteomics 18, e1700436 (2018).
Bollineni, R. C., Koehler, C. J., Gislefoss, R. E., Anonsen, J. H. & Thiede, B. Large-scale intact glycopeptide identification by Mascot database search. Sci. Rep. 8, 2117 (2018).
Nasir, W. et al. SweetNET: a bioinformatics workflow for glycopeptide MS/MS spectral analysis. J. Proteome Res. 15, 2826–2840 (2016).
Park, G. W. et al. Integrated GlycoProteome analyzer (I-GPA) for automated identification and quantitation of site-specific N-glycosylation. Sci. Rep. 6, 21175 (2016).
Liu, G. et al. A comprehensive, open-source platform for mass spectrometry-based glycoproteomics data analysis. Mol. Cell. Proteom. 16, 2032–2047 (2017).
Maxwell, E. et al. GlycReSoft: a software package for automated recognition of glycans from LC/MS data. PLoS ONE 7, e45474 (2012).
Pioch, M., Hoffmann, M., Pralow, A., Reichl, U. & Rapp, E. glyXtool(MS): an open-source pipeline for semiautomated analysis of glycopeptide mass spectrometry data. Anal. Chem. 90, 11908–11916 (2018).
Bern, M., Kil, Y. J. & Becker, C. Byonic: advanced peptide and protein identification software. Curr. Protoc. Bioinformatics https://doi.org/10.1002/0471250953.bi1320s40 (2012).
Go, E. P. et al. The opportunity cost of automated glycopeptide analysis: case study profiling the SARS-CoV-2 S glycoprotein. Anal. Bioanal. Chem. 413, 7215–7227 (2021).
Trinidad, J. C., Schoepfer, R., Burlingame, A. L. & Medzihradszky, K. F. N- and O-glycosylation in the murine synaptosome. Mol. Cell Proteom. 12, 3474–3488 (2013).
Medzihradszky, K. F., Kaasik, K. & Chalkley, R. J. Tissue-specific glycosylation at the glycopeptide level. Mol. Cell. Proteom. 14, 2103–2110 (2015).
Zhang, R., Zhu, J., Lubman, D. M., Mechref, Y. & Tang, H. GlycoHybridSeq: automated identification of N-linked glycopeptides using electron transfer/high-energy collision dissociation (EThcD). J. Proteome Res. 20, 3345–3352 (2021).
Pompach, P., Chandler, K. B., Lan, R., Edwards, N. & Goldman, R. Semi-automated identification of N-glycopeptides by hydrophilic interaction chromatography, nano-reverse-phase LC-MS/MS, and glycan database search. J. Proteome Res. 11, 1728–1740 (2012).
He, L., Xin, L., Shan, B., Lajoie, G. A. & Ma, B. GlycoMaster DB: software to assist the automated identification of N-linked glycopeptides by tandem mass spectrometry. J. Proteome Res. 13, 3881–3895 (2014).
Schulze, S. et al. SugarPy facilitates the universal, discovery-driven analysis of intact glycopeptides. Bioinformatics https://doi.org/10.1093/bioinformatics/btaa1042 (2020).
Polasky, D. A., Yu, F., Teo, G. C. & Nesvizhskii, A. I. Fast and comprehensive N- and O-glycoproteomics analysis with MSFragger-Glyco. Nat. Methods 17, 1125–1132 (2020).
Lu, L., Riley, N. M., Shortreed, M. R., Bertozzi, C. R. & Smith, L. M. O-Pair search with MetaMorpheus for O-glycopeptide characterization. Nat. Methods 17, 1133–1138 (2020).
Lynn, K. S. et al. MAGIC: an automated N-linked glycoprotein identification tool using a Y1-ion pattern matching algorithm and in silico MS(2) approach. Anal. Chem. 87, 2466–2473 (2015).
Toghi Eshghi, S., Shah, P., Yang, W., Li, X. & Zhang, H. GPQuest: a spectral library matching algorithm for site-specific assignment of tandem mass spectra to intact N-glycopeptides. Anal. Chem. 87, 5181–5188 (2015).
Yang, Y. et al. GproDIA enables data-independent acquisition glycoproteomics with comprehensive statistical control. Nat. Commun. 12, 6073 (2021).
Zhu, H., Qiu, C., Gryniewicz-Ruzicka, C. M., Keire, D. A. & Ye, H. Multiplexed comparative analysis of intact glycopeptides using electron-transfer dissociation and synchronous precursor selection based triple-stage mass spectrometry. Anal. Chem. 92, 7547–7555 (2020).
Caval, T., Zhu, J. & Heck, A. J. R. Simply extending the mass range in electron transfer higher energy collisional dissociation increases confidence in N-glycopeptide identification. Anal. Chem. 91, 10401–10406 (2019).
Zeng, W. F., Cao, W. Q., Liu, M. Q., He, S. M. & Yang, P. Y. Precise, fast and comprehensive analysis of intact glycopeptides and modified glycans with pGlyco3. Nat. Methods 18, 1515–1523 (2021).
Kawahara, R. et al. The complexity and dynamics of the tissue glycoproteome associated with prostate cancer progression. Mol. Cell Proteom. 20, 100026 (2021).
Sanda, M., Benicky, J. & Goldman, R. Low collision energy fragmentation in structure-specific glycoproteomics analysis. Anal. Chem. 92, 8262–8267 (2020).
Chalkley, R. J. & Baker, P. R. Use of a glycosylation site database to improve glycopeptide identification from complex mixtures. Anal. Bioanal. Chem. 409, 571–577 (2017).
Zeng, W. F. et al. pGlyco: a pipeline for the identification of intact N-glycopeptides by using HCD- and CID-MS/MS and MS3. Sci. Rep. 6, 25102 (2016).
Shen, J. et al. StrucGP: de novo structural sequencing of site-specific N-glycan on glycoproteins using a modularization strategy. Nat. Methods 18, 921–929 (2021).
Polasky, D. A., Geiszler, D. J., Fengchao, Y. & Nesvizhkii, A. I. Multi-attribute glycan identification and FDR control for glycoproteomics. Mol. Cell. Proteom. 21, 100205 (2022).
Yu, J. et al. Distinctive MS/MS fragmentation pathways of glycopeptide-generated oxonium ions provide evidence of the glycan structure. Chemistry 22, 1114–1124 (2016).
Halim, A. et al. Assignment of saccharide identities through analysis of oxonium ion fragmentation profiles in LC-MS/MS of glycopeptides. J. Proteome Res. 13, 6024–6032 (2014). Critical work demonstrating that oxonium fragment ions can be used to distinguish isobaric GlcNAc and GalNAc glycosylation events.
Toghi Eshghi, S. et al. Classification of tandem mass spectra for identification of N- and O-linked glycopeptides. Sci. Rep. 6, 37189 (2016).
Pett, C. et al. Effective assignment of alpha2,3/alpha2,6-sialic acid isomers by LC-MS/MS-based glycoproteomics. Angew. Chem. Int. Ed. 57, 9320–9324 (2018).
Pap, A., Tasnadi, E., Medzihradszky, K. F. & Darula, Z. Novel O-linked sialoglycan structures in human urinary glycoproteins. Mol. Omics 16, 156–164 (2020).
Park, G. W. et al. Classification of mucin-type O-glycopeptides using higher-energy collisional dissociation in mass spectrometry. Anal. Chem. 92, 9772–9781 (2020).
Jeong, H. K. et al. Computational classification of core and outer fucosylation of N-glycoproteins in human plasma using collision-induced dissociation in mass spectrometry. Rapid Commun. Mass Spectrom. 34, e8917 (2020).
Hwang, H. et al. Machine learning classifies core and outer fucosylation of N-glycoproteins using mass spectrometry. Sci. Rep. 10, 318 (2020).
Dang, L. et al. Recognition of bisecting N-glycans on intact glycopeptides by two characteristic ions in tandem mass spectra. Anal. Chem. 91, 5478–5482 (2019).
Chalkley, R. J., Medzihradszky, K. F., Darula, Z., Pap, A. & Baker, P. R. The effectiveness of filtering glycopeptide peak list files for Y ions. Mol. Omics 16, 147–155 (2020).
Lee, H. K. et al. Selective identification of alpha-galactosyl epitopes in N-glycoproteins using characteristic fragment ions from higher-energy collisional dissociation. Anal. Chem. 92, 13144–13154 (2020).
Zhu, H. et al. Identifying sialylation linkages at the glycopeptide level by glycosyltransferase labeling assisted mass spectrometry (GLAMS). Anal. Chem. 92, 6297–6303 (2020).
Wen, L. et al. A one-step chemoenzymatic labeling strategy for probing sialylated Thomsen-Friedenreich antigen. ACS Cent. Sci. 4, 451–457 (2018).
You, X. et al. Chemoenzymatic approach for the proteomics analysis of mucin-type core-1 O-glycosylation in human serum. Anal. Chem. 90, 12714–12722 (2018).
Yang, S., Wu, W. W., Shen, R. F., Bern, M. & Cipollo, J. Identification of sialic acid linkages on intact glycopeptides via differential chemical modification using IntactGIG-HILIC. J. Am. Soc. Mass Spectrom. 29, 1273–1283 (2018).
Elias, J. E. & Gygi, S. P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 (2007).
Kawahara, R. et al. Community evaluation of glycoproteomics informatics solutions reveals high-performance search strategies for serum glycopeptide analysis. Nat. Methods 18, 1304–1316 (2021).
Zhu, Z., Su, X., Go, E. P. & Desaire, H. New glycoproteomics software, GlycoPep evaluator, generates decoy glycopeptides de novo and enables accurate false discovery rate analysis for small data sets. Anal. Chem. 86, 9212–9219 (2014).
Thaysen-Andersen, M. et al. Human neutrophils secrete bioactive paucimannosidic proteins from azurophilic granules into pathogen-infected sputum. J. Biol. Chem. 290, 8789–8802 (2015).
Chen, Z. et al. In-depth site-specific analysis of N-glycoproteome in human cerebrospinal fluid and glycosylation landscape changes in Alzheimer’s disease. Mol. Cell. Proteom. 20, 100081 (2021).
Sinha, A. et al. N-glycoproteomics of patient-derived xenografts: a strategy to discover tumor-associated proteins in high-grade serous ovarian cancer. Cell Syst. 8, 345–351.e344 (2019).
Goyallon, A., Cholet, S., Chapelle, M., Junot, C. & Fenaille, F. Evaluation of a combined glycomics and glycoproteomics approach for studying the major glycoproteins present in biofluids: application to cerebrospinal fluid. Rapid Commun. Mass Spectrom. 29, 461–473 (2015).
Stadlmann, J. et al. Comparative glycoproteomics of stem cells identifies new players in ricin toxicity. Nature 549, 538–542 (2017).
Zhang, Q., Ma, C., Chin, L. S. & Li, L. Integrative glycoproteomics reveals protein N-glycosylation aberrations and glycoproteomic network alterations in Alzheimer’s disease. Sci. Adv. 6, eabc5802 (2020).
Ji, Y. et al. Integrated proteomic and N-glycoproteomic analyses of doxorubicin sensitive and resistant ovarian cancer cells reveal glycoprotein alteration in protein abundance and glycosylation. Oncotarget 8, 13413–13427 (2017).
Li, Q. K. et al. An integrated proteomic and glycoproteomic approach uncovers differences in glycosylation occupancy from benign and malignant epithelial ovarian tumors. Clin. Proteom. 14, 16 (2017).
Hu, Y. et al. Integrated proteomic and glycoproteomic characterization of human high-grade serous ovarian carcinoma. Cell Rep. 33, 108276 (2020).
Mereiter, S., Balmana, M., Campos, D., Gomes, J. & Reis, C. A. Glycosylation in the era of cancer-targeted therapy: where are we heading? Cancer Cell 36, 6–16 (2019).
Wang, Z. et al. Integrated proteomic and N-glycoproteomic analyses of human breast cancer. J. Proteome Res. 19, 3499–3509 (2020).
Zhou, Y. et al. Proteomic analysis of the air-way fluid in lung cancer. Detection of periostin in bronchoalveolar lavage (BAL). Front. Oncol. 10, 1072 (2020).
Li, X. et al. In-depth analysis of secretome and N-glycosecretome of human hepatocellular carcinoma metastatic cell lines shed light on metastasis correlated proteins. Oncotarget 7, 22031–22049 (2016).
Cima, I. et al. Cancer genetics-guided discovery of serum biomarker signatures for diagnosis and prognosis of prostate cancer. Proc. Natl Acad. Sci. USA 108, 3342–3347 (2011).
Sudhir, P. R. et al. Label-free quantitative proteomics and N-glycoproteomics analysis of KRAS-activated human bronchial epithelial cells. Mol. Cell. Proteom. 11, 901–915 (2012).
Yu, Q. et al. Targeted mass spectrometry approach enabled discovery of O-glycosylated insulin and related signaling peptides in mouse and human pancreatic islets. Anal. Chem. 89, 9184–9191 (2017).
Malaker, S. A. et al. Revealing the human mucinome. Preprint at bioRxiv https://doi.org/10.1101/2021.01.27.428510 (2021).
Zilmer, M. et al. Novel congenital disorder of O-linked glycosylation caused by GALNT2 loss of function. Brain 143, 1114–1126 (2020).
Schjoldager, K. T. et al. Probing isoform-specific functions of polypeptide GalNAc-transferases using zinc finger nuclease glycoengineered SimpleCells. Proc. Natl Acad. Sci. USA 109, 9893–9898 (2012).
Stavenhagen, K. et al. Tumor cells express pauci- and oligomannosidic N-glycans in glycoproteins recognized by the mannose receptor (CD206). Cell Mol. Life Sci. 78, 5569–5585 (2021).
Pirro, M. et al. Characterization of macrophage galactose-type lectin (MGL) ligands in colorectal cancer cell lines. Biochim. Biophys. Acta Gen. Subj. 1864, 129513 (2020).
Pirro, M. et al. Glycoproteomic analysis of MGL-binding proteins on acute T-cell leukemia cells. J. Proteome Res. 18, 1125–1132 (2019).
Barefoot, M. E. et al. Multi-omic pathway and network analysis to identify biomarkers for hepatocellular carcinoma. Annu. Int. Conf. IEEE Eng. Med. Biol. Soc. 2019, 1350–1354 (2019).
Cao, L. et al. Integrating transcriptomics, proteomics, glycomics and glycoproteomics to characterize paclitaxel resistance in breast cancer cells. J. Proteom. 243, 104266 (2021).
Rolland, D. C. M. et al. Functional proteogenomics reveals biomarkers and therapeutic targets in lymphomas. Proc. Natl Acad. Sci. USA 114, 6581–6586 (2017).
Sudhir, P. R. et al. Integrative omics connects N-glycoproteome-wide alterations with pathways and regulatory events in induced pluripotent stem cells. Sci. Rep. 6, 36109 (2016).
Park, J. M. et al. Integrated analysis of global proteome, phosphoproteome, and glycoproteome enables complementary interpretation of disease-related protein networks. Sci. Rep. 5, 18189 (2015).
Luo, B. et al. Bifunctional magnetic covalent organic framework for simultaneous enrichment of phosphopeptides and glycopeptides. Anal. Chim. Acta 1177, 338761 (2021).
Kang, T. et al. Characterization of signaling pathways associated with pancreatic beta-cell adaptive flexibility in compensation of obesity-linked diabetes in db/db mice. Mol. Cell. Proteom. 19, 971–993 (2020).
Yang, R. et al. Integrated proteomic, phosphoproteomic and N-glycoproteomic analyses of chicken eggshell matrix. Food Chem. 330, 127167 (2020).
Kawahara, R. et al. Integrated proteomics reveals apoptosis-related mechanisms associated with placental malaria. Mol. Cell. Proteom. 18, 182–199 (2019).
Cao, L. et al. Proteogenomic characterization of pancreatic ductal adenocarcinoma. Cell 184, 5031–5052.e5026 (2021).
Woods, R. J. Predicting the structures of glycans, glycoproteins, and their complexes. Chem. Rev. 118, 8005–8024 (2018).
Bagdonas, H., Fogarty, C. A., Fadda, E. & Agirre, J. The case for post-predictional modifications in the AlphaFold protein structure database. Nat. Struct. Mol. Biol. 28, 869–870 (2021).
Joshi, H. J. et al. GlycoDomainviewer: a bioinformatics tool for contextual exploration of glycoproteomes. Glycobiology 28, 131–136 (2018).
Cioce, A. et al. Cell-specific bioorthogonal tagging of glycoproteins. Preprint at bioRxiv https://doi.org/10.1101/2021.07.28.454135 (2021).
Poulos, R. C. et al. Strategies to enable large-scale proteomics for reproducible research. Nat. Commun. 11, 3793 (2020).
Wada, Y. et al. Comparison of the methods for profiling glycoprotein glycans–HUPO human disease glycomics/proteome initiative multi-institutional study. Glycobiology 17, 411–422 (2007).
Wada, Y. et al. Comparison of methods for profiling O-glycosylation: human proteome organisation human disease glycomics/proteome initiative multi-institutional study of IgA1. Mol. Cell. Proteom. 9, 719–727 (2010).
Leymarie, N. et al. Interlaboratory study on differential analysis of protein glycosylation by mass spectrometry: the ABRF glycoprotein research multi-institutional study 2012. Mol. Cell. Proteom. 12, 2935–2951 (2013).
De Leoz, M. L. A. et al. NIST interlaboratory study on glycosylation analysis of monoclonal antibodies: comparison of results from diverse analytical methods. Mol. Cell. Proteom. 19, 11–30 (2020).
Wilkinson, M. D. et al. The FAIR guiding principles for scientific data management and stewardship. Sci. Data 3, 160018 (2016).
York, W. S. et al. MIRAGE: the minimum information required for a glycomics experiment. Glycobiology 24, 402–406 (2014).
Watanabe, Y., Aoki-Kinoshita, K. F., Ishihama, Y. & Okuda, S. GlycoPOST realizes FAIR principles for glycomics mass spectrometry data. Nucleic Acids Res. 49, D1523–D1528 (2021).
Rojas-Macias, M. A. et al. Towards a standardized bioinformatics infrastructure for N- and O-glycomics. Nat. Commun. 10, 3275 (2019).
Yamada, I. et al. The GlyCosmos Portal: a unified and comprehensive web resource for the glycosciences. Nat. Methods 17, 649–650 (2020).
Perez-Riverol, Y., Alpi, E., Wang, R., Hermjakob, H. & Vizcaino, J. A. Making proteomics data accessible and reusable: current state of proteomics databases and repositories. Proteomics 15, 930–949 (2015).
Perez-Riverol, Y. et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47, D442–D450 (2019).
Takahashi, K. et al. Naturally occurring structural isomers in serum IgA1 O-glycosylation. J. Proteome Res. 11, 692–702 (2012).
Akune, Y. et al. Comprehensive analysis of the N-glycan biosynthetic pathway using bioinformatics to generate UniCorn: a theoretical N-glycan structure database. Carbohydr. Res. 431, 56–63 (2016).
McDonald, A. G., Tipton, K. F. & Davey, G. P. A knowledge-based system for display and prediction of O-glycosylation network behaviour in response to enzyme knockouts. PLoS Comput. Biol. 12, e1004844 (2016).
Kolarich, D. et al. Glycoproteomic characterization of butyrylcholinesterase from human plasma. Proteomics 8, 254–263 (2008).
Wang, S. et al. Synthesis of rhamnosylated arginine glycopeptides and determination of the glycosidic linkage in bacterial elongation factor P. Chem. Sci. 8, 2296–2302 (2017).
Loke, I., Ostergaard, O., Heegaard, N. H. H., Packer, N. H. & Thaysen-Andersen, M. Paucimannose-rich N-glycosylation of spatiotemporally regulated human neutrophil elastase modulates its immune functions. Mol. Cell. Proteom. 16, 1507–1527 (2017).
Halim, A. et al. Site-specific characterization of threonine, serine, and tyrosine glycosylations of amyloid precursor protein/amyloid beta-peptides in human cerebrospinal fluid. Proc. Natl Acad. Sci. USA 108, 11848–11853 (2011).
Pan, J. et al. Glycoproteomics-based signatures for tumor subtyping and clinical outcome prediction of high-grade serous ovarian cancer. Nat. Commun. 11, 6139 (2020).
Larsson, J. M. et al. Altered O-glycosylation profile of MUC2 mucin occurs in active ulcerative colitis and is associated with increased inflammation. Inflamm. Bowel Dis. 17, 2299–2307 (2011).
Klaric, L. et al. Glycosylation of immunoglobulin G is regulated by a large network of genes pleiotropic with inflammatory diseases. Sci. Adv. 6, eaax0301 (2020).
Lu, L. L. et al. A functional role for antibodies in tuberculosis. Cell 167, 433–443.e414 (2016).
Offersen, R. et al. HIV antibody Fc N-linked glycosylation is associated with viral rebound. Cell Rep. 33, 108502 (2020).
Ugonotti, J. et al. N-Acetyl-β-d-hexosaminidases mediate the generation of paucimannosidic proteins via a putative non-canonical truncation pathway in human neutrophils. Glycobiology 32, 218–229 (2022).
Noach, I. et al. Recognition of protein-linked glycans as a determinant of peptidase activity. Proc. Natl Acad. Sci. USA 114, E679–E688 (2017).
Pluvinage, B. et al. Architecturally complex O-glycopeptidases are customized for mucin recognition and hydrolysis. Proc. Natl Acad. Sci. USA 118, e2019220118 (2021).
McKitrick, T. R. et al. Development of smart anti-glycan reagents using immunized lampreys. Commun. Biol. 3, 91 (2020).
John, A. et al. Yeast- and antibody-based tools for studying tryptophan C-mannosylation. Nat. Chem. Biol. 17, 428–437 (2021).
Chua, X. Y. et al. Tandem mass tag approach utilizing pervanadate BOOST channels delivers deeper quantitative characterization of the tyrosine phosphoproteome. Mol. Cell. Proteom. 19, 730–743 (2020).
Budnik, B., Levy, E., Harmange, G. & Slavov, N. SCoPE-MS: mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biol. 19, 161 (2018).
Kafader, J. O. et al. Multiplexed mass spectrometry of individual ions improves measurement of proteoforms and their complexes. Nat. Methods 17, 391–394 (2020).
Kafader, J. O. et al. Individual ion mass spectrometry enhances the sensitivity and sequence coverage of top-down mass spectrometry. J. Proteome Res. 19, 1346–1350 (2020).
Worner, T. P. et al. Resolving heterogeneous macromolecular assemblies by Orbitrap-based single-particle charge detection mass spectrometry. Nat. Methods 17, 395–398 (2020).
Minoshima, F., Ozaki, H., Odaka, H. & Tateno, H. Integrated analysis of glycan and RNA in single cells. iScience 24, 102882 (2021).
Restrepo-Perez, L., Wong, C. H., Maglia, G., Dekker, C. & Joo, C. Label-free detection of post-translational modifications with a nanopore. Nano Lett. 19, 7957–7964 (2019).
Swearingen, K. E. et al. Interrogating the plasmodium sporozoite surface: identification of surface-exposed proteins and demonstration of glycosylation on CSP and TRAP by mass spectrometry-based proteomics. PLoS Pathog. 12, e1005606 (2016).
Hardiville, S. et al. TATA-box binding protein O-GlcNAcylation at T114 regulates formation of the B-TFIID complex and is critical for metabolic gene regulation. Mol. Cell 77, 1143–1152.e1147 (2020).
Sharapov, S. Z. et al. Replication of 15 loci involved in human plasma protein N-glycosylation in 4802 samples from four cohorts. Glycobiology 31, 82–88 (2021).
AlQuraishi, M. & Sorger, P. K. Differentiable biology: using deep learning for biophysics-based and data-driven modeling of molecular mechanisms. Nat. Methods 18, 1169–1180 (2021).
Greener, J. G., Kandathil, S. M., Moffat, L. & Jones, D. T. A guide to machine learning for biologists. Nat. Rev. Mol. Cell Biol. 23, 40–55 (2022).
Meyer, J. G. Deep learning neural network tools for proteomics. Cell Rep. Methods 1, 100003 (2021).
Taus, T. et al. Universal and confident phosphorylation site localization using phosphoRS. J. Proteome Res. 10, 5354–5362 (2011).
Segu, Z. M. & Mechref, Y. Characterizing protein glycosylation sites through higher-energy C-trap dissociation. Rapid Commun. Mass Spectrom. 24, 1217–1225 (2010).
Caval, T. et al. Targeted analysis of lysosomal directed proteins and their sites of mannose-6-phosphate modification. Mol. Cell. Proteom. 18, 16–27 (2019).
Varki, A. et al. Symbol nomenclature for graphical representations of glycans. Glycobiology 25, 1323–1324 (2015).
Neelamegham, S. et al. Updates to the symbol nomenclature for glycans guidelines. Glycobiology 29, 620–624 (2019).
Acknowledgements
N.E.S. is supported by an Australian Research Council Future Fellowship (FT200100270). B.L.P. is supported by an Australian National Health and Medical Research Emerging Leader Grant (APP 2009642). M.T.-A. is supported by an Australian Research Council Future Fellowship (FT210100455). D.A.P. and A.I.N. are supported by NIH grants R01-GM-094231 and U24-CA210967. S.A.M. is supported by the Yale Science Development Fund and the Yale SEAS/Science Program to Advance Research Collaboration (SPARC). A.H. is supported by a research grant (00025438) from VILLUM FONDEN. K.S. is supported by an Excellence grant from the Novo Nordisk Foundation (NNF17OC0026030). H.H.W. is supported by the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (GlycoSkin H2020-ERC; 772735), Lundbeck Foundation (R313-2019-869) and the Neye Foundation. The Copenhagen Center for Glycomics including I.B., K.S., A.H., H.H.W. and S.Y.V. are supported by a Danish National Research Foundation grant (DNRF107). N.M.R. acknowledges support from a NIH Predoctoral to Postdoctoral Transition award (K00 CA212454). K.F.A.-K. is supported by the Japan Science and Technology Agency (JST) and National Bioscience Database Center (NBDC) of Japan, grant number 17934031.
Author information
Authors and Affiliations
Contributions
Introduction (B.L.P. and M.T.-A.), Experimentation (I.B., S.A.M., D.A.P., N.M.R., K.S., A.I.N., C.R.B. and H.H.W.), Results (S.Y.V., A.H., D.A.P. and S.A.M.), Applications (I.B., K.S., C.R.B. and H.H.W.), Reproducibility and data deposition (K.F.A.-K. and M.T.-A.), Limitations and optimizations (N.E.S.), Outlook (N.E.S. and B.L.P.). Overview of the Primer (N.E.S.).
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Methods Primers thanks Yehia Mechref and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
Reporting guidelines for glycomics: https://www.beilstein-institut.de/en/projects/mirage/guidelines
Glossary
- Non-templated
-
A process that is not guided by a template, in contrast to templated processes such as DNA transcription and translation.
- Anomeric linkages
-
A linkage description that denotes the connectivity of one monosaccharide to another relative to the anomeric centre of the monosaccharide.
- Intact glycopeptides
-
Glycopeptides decorated with their native or near-native glycoforms.
- Bottom-up glycoproteomics
-
The analysis of glycoproteome samples based on the identification of proteolytically generated glycopeptides.
- Proteoforms
-
A proteoform is a specific form of a protein defined by its exact amino acid sequence and post-translational modification status at specific residues in the protein.
- Dynamic range
-
Within the context of bottom-up glycoproteomics, the range of quantifiable ion intensities between the most abundant observable glycopeptide and the least abundant glycopeptide.
- Glycoforms
-
Different molecular forms of a glycoprotein for which either the glycan composition or site of attachment can differ from another glycosylated form of the protein.
- Chaotropic agents
-
Agents that cause perturbations in non-covalent forces, leading to the disruption of molecular structures aiding in the solubilization of proteins.
- Positive polarity mode
-
A mode of operation of a mass spectrometer conducive to the analysis of positively charged ions.
- Proteome coverage
-
The depth of analysis achieved for a given proteome experiment, referring to the total number of protein identifications or the average number of peptides or sequence coverage identified per protein.
- Exoprotease
-
A protease that catalyses the removal of N-terminal or C-terminal amino acids from a protein sequence.
- Endoprotease
-
A protease that catalyses the cleavage of amino acids within a protein sequence.
- Non-reducing termini
-
The termini of a carbohydrate chain that are unable to initiate a reduction reaction.
- Lectin
-
A general term given to describe carbohydrate-binding proteins, which typically have affinity for specific sugars and modest binding affinities.
- Click chemistry
-
A loosely defined set of chemical conjugation reactions that are typically highly efficient and occur between small chemical groups such as alkyne and azide groups.
- Bump-and-hole strategy
-
A chemical biology approach that involves the modification of enzymes to accommodate a non-native small molecule, such as a nucleotide-linked carbohydrate containing a chemical handle, enabling enrichment or imaging.
- Isomeric glycoforms
-
Glycans that have the same elemental composition but differ in monosaccharide arrangement; also isobaric in nature by definition.
- Online glycopeptide separation
-
The separation of glycopeptides or peptides using chromatographic approaches whereby the eluting samples are directly introduced into a mass spectrometer.
- Offline separation
-
Separations of glycopeptides or peptides using chromatographic approaches not directly interfaced with a mass spectrometer.
- Cross-ring fragments
-
Fragment ions that result from cleavage across a carbohydrate ring.
- Secondary gas-phase structure effects
-
The reduced efficiency of fragmentation caused by the formation of secondary structure within the gas phase of a polypeptide backbone.
- Isotopologues
-
Chemical species that differ in the number of neutrons resulting from the incorporation of stable isotopes such as 13C, 15N, 18O or 2H; these chemical species have near-identical physicochemical properties, but are observed with a heavier mass using mass spectrometry
- Sequon
-
A sequence (motif) of amino acids that corresponds to a predictable site of glycosylation.
- Top-down glycoproteomics
-
The analysis of glycoproteome samples based on intact, undigested glycoprotein species.
Rights and permissions
About this article

Cite this article
Bagdonaite, I., Malaker, S.A., Polasky, D.A. et al. Glycoproteomics. Nat Rev Methods Primers 2, 48 (2022). https://doi.org/10.1038/s43586-022-00128-4
Accepted:
Published:
DOI: https://doi.org/10.1038/s43586-022-00128-4
This article is cited by
-
Thermophoretic glycan profiling of extracellular vesicles for triple-negative breast cancer management
Nature Communications (2024)
-
Prediction of glycopeptide fragment mass spectra by deep learning
Nature Communications (2024)
-
A high-throughput workflow for comprehensive glycopeptide mapping
Nature Biomedical Engineering (2023)
-
Protein cysteine S-glycosylation: oxidative hydrolysis of protein S-glycosidic bonds in aqueous alkaline environments
Amino Acids (2023)
-
Recent trends in glycoproteomics by characterization of intact glycopeptides
Analytical and Bioanalytical Chemistry (2023)
