
Abstract
The surface ocean is an important source of longer-lived volatile organic iodine compounds, such as methyl and ethyl iodide. However, their production in deep waters is poorly constrained. Here, we combine shipborne seawater and sediment core analyses to investigate the temporal and vertical distribution of methyl and ethyl iodide concentrations in the Bering and Chukchi Seas in July 2017 and 2018, and Funka Bay, Japan, between March 2018 and December 2019. We find the highest methyl and ethyl iodide concentrations in sediment porewaters and at the sediment surface. In Funka Bay, concentrations in the sediment and bottom waters strongly increased following the spring bloom and subsequent settling of phytodetritus to the seafloor. Dark incubation experiments suggest that diatom aggregates are a source of methyl and ethyl iodide to the ambient water. Our findings suggest that phytodetritus at the seafloor is a potential source of volatile organic iodine compounds to the water column in polar and subpolar coastal oceans.
Similar content being viewed by others
Introduction
The ocean surface is the dominant source of atmospheric iodine1,2. Volatile organic iodine compounds (VOIs) such as methyl iodide (CH3I) contribute to 20–40% of supply of the reactive iodine in the upper troposphere and lower stratosphere3. Although another longer-lived VOI, C2H5I, is less abundant in air and surface seawater worldwide4,5,6,7, first estimates suggest that sea-to-air flux of C2H5I in the world coastal ocean accounts for 10% of total monohaloalkane iodine emission8.
In the ocean the dominant producers of VOIs have been thought to be marine plants such as macro algae9,10, micro algae11,12,13, and photosynthetic bacteria14,15. Very few studies have taken notice of VOI production by heterotrophic bacteria16,17,18. Abiotic photochemical production of CH3I has been suggested19. Most previous studies have focused on the production of VOIs in the upper euphotic zone, where marine plants grow. High levels of CH3I in surface water occur widely in the oligotrophic subtropical open oceans, and relatively low levels occur in subpolar and polar open oceans6. In contrast, concentrations of C2H5I in surface water are high in subpolar and polar continental shelf areas compared with the substantially lower levels in subtropical open oceans6,20. Recent studies have shown high concentrations of C2H5I and some VOIs in bottom water of the polar Chukchi Sea and northern Bering Sea21, and subpolar Funka Bay, Japan22. These high concentrations have given rise to the question of whether there is a VOI source near the sea floor. In this study, we measure the concentrations of VOIs in the sea-floor sediment by shipboard observations (Fig. 1). We also conducted laboratory incubations to test the hypothesis that diatom aggregates settled on the sea floor emit VOIs in darkness.

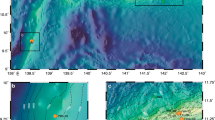
a Map of North Pacific including study areas. b Map of Hokkaido Island in Japan. c Sampling site of Funka Bay in Hokkaido, Japan. Black circle indicates the station 30. d Sampling sites of Chukchi and Bering Seas. White and gray circles indicate sampling sites in 2017 and 2018, respectively. Black circle indicates a common sampling site in 2017 and 2018. Northern Bering Sea stations and Bering Sea stations are 14, 19, 22 and 4, 8, 21, 23 respectively. Video recordings of the sea floor were taken at station 30 in Funka Bay and at stations 4, 8, 14, and 22 in the Bering Sea (Supplementary Movie 1–4, 6).
Results and discussion
Temporal variations of VOIs profile in water column and sediment in Funka Bay
Time-series cross sections of chlorophyll-a, C2H5I, and CH3I in the water column and sediment in Funka Bay, Hokkaido, Japan, (Fig. 2) indicate a potential source of VOIs in the sea-floor sediment. All observation data are provided in Supplementary Data 1–4. The annual maximum chlorophyll-a concentration (>12 μg L–1) in the surface water was found in March of both 2018 and 2019 (Fig. 2a). Such high levels of chlorophyll-a indicate a spring diatom bloom in the bay23. The maximum level of 29.7 μg L–1 on 4 March 2019 was much higher than 12.5 μg L–1 on 18 March 2018. It suggests that the magnitude of diatom bloom in 2019 was greater than 2018. From microscopic image analysis, Thalassiosira nordenskioeldii occupied 14.2% of number of phytoplankton cells (n = 1209) collected by plankton net (mesh = 100 μm) towing from the bottom layer (87 m depth) to the surface on 15 March 2019. Other dominant species were Chaetoceros spp. and other Thalassiosira sp. Although small-sized phytoplanktons could not be detected by the microscopic analysis, we considered that diatoms were the dominant species in the spring bloom 2019 on the ground of the pigment analysis result that diatoms occupied >90% of depth integrated chl-a within euphotic layer in spring blooms 2010 and 2011 in the Funka Bay24. In April of both years the concentrations dropped below 4 μg L–1, indicating termination of the spring bloom. In the bottom water (80–96 m) we found high concentrations of chlorophyll-a (>7 μg L–1) from the beginning of the diatom bloom in early March of both years until middle of March 2018 and April 2019. The bottom water was below the euphotic zone, so the chlorophyll-a maximum in the dark bottom water was caused by settling of diatom aggregates produced in the surface water during the bloom. The concentration of C2H5I in the surface sediment (Fig. 2b) showed annual minima on 4 March 2018 (25 pmolL–1) and 14 December 2018 (24 pmolL–1), and dramatically increased on 18 March 2018 (257 pmol L–1) and 15 March 2019 (178 pmol L–1). These increases were found 11–14 d after the detections of increases in bottom-water chlorophyll-a. The high concentrations of C2H5I in the sediment continued through August and dropped in October. In the water column, the annual minimum for C2H5I (<1 pmolL–1) was during the winter and into the spring bloom. The bottom-water concentrations dramatically increased post-bloom on 8 April 2018 (5.4 pmolL–1) and 14 April 2019 (5.4 pmolL–1), 16–30 d after the detections of dramatic increases in the sediment. The high concentrations in the bottom water lasted until August in 2018 and until June in 2019.

a Chl-a in water column. b C2H5I in water column and sediment. c CH3I in water column and sediment. The dates of annual rises in concentration are marked with triangles below each panel. Solid black lines indicate the euphotic zone depth, at which photosynthetically active radiation (PAR) is 1% of the surface PAR. White dotted lines separate bottom water below 80 m depth. Squares outlined with black-and-white dotted lines indicate the temporal-vertical range where C2H5I and CH3I concentrations decreased with time.
As for CH3I, the timings of increases in the sediment and bottom water (Fig. 2c) were consistent with those of C2H5I. The annual average concentrations of surface-sediment C2H5I (118 pmol L–1) and CH3I (51 pmol L–1) were 20 and 8 times those in the bottom water, respectively. It is therefore obvious that the increases in bottom water were led by transfer from much higher concentrations in the surface sediment. The production of C2H5I and CH3I from diatom aggregates in relation to bacterial activity in darkness has already been reported18. We, therefore, assumed that the C2H5I and CH3I in the sediment originates from diatom aggregates settled on the sea floor. This assumption was confirmed by dark incubation experiments using diatom aggregates collected in Funka Bay.
At the lower part of middle layer water (65–75 m) and bottom layer water (85 m), the concentrations of C2H5I and CH3I decreased by half from 12 June to 29 August in 2019 (squares outlined with black-and-white dotted lines in Fig. 2b, c), where the water mass25 (W or WO waters) remained at the layers between the two dates (Supplementary Fig. S1). However, elevated concentrations of phosphate (PO43−) at the layers, which were derived from decomposition of organic matter settled on the sea floor, decreased by 0–17% only between the two dates (Supplementary Fig. S2). As for the obvious decreases in C2H5I and CH3I concentrations at the layers, we presumed that biological losses of C2H5I and CH3I occurred in the water. In 2018, elevations of C2H5I concentration and following decreases at 65–75-m depths were found between May and June, however obvious elevation of CH3I concentration at the depths were not detected. It is possible that biological losses of C2H5I and CH3I in the middle layer had advanced in 2018 compared to 2019.
The biological loss rate for CH3I has been reported to be 9% day−1 at 7 °C in the subarctic Atlantic Ocean, where chemical loss is negligible because of low temperature26. The net loss rates (loss—supply) of our study were 2% day−1 (= Δ1.61 pmol L−1 / 68 days) for CH3I and 0.5% day−1 (= Δ1.19 pmol L−1 / 68 days) for C2H5I at the layer between 12 June and 29 August (68 days interval). The lower rates of our study would be due to consecutive supplies of CH3I and C2H5I from the sediment source. We considered that the biological losses in water column near the bottom inhibited the transport of sediment source CH3I and C2H5I to the surface mixed layer and air. And thus, the sediment source CH3I and C2H5I would have a small impact on the VOIs emission to the air, although we will conclude that phytodetritus settled on the sea floor is a hotspot of VOIs production in the ocean.
Here, we briefly mention the obvious CH3I maxima, which were found from May to August within the oligotrophic euphotic zone (Fig. 2c). While the chlorophyll-a concentrations in that period were much lower than those in the bloom, CH3I concentrations (4.5–8.5 pmol L–1) in May–August were approximately five times those in the bloom. In contrast, C2H5I concentrations (0.6–1.7 pmol L–1) in the euphotic zone during the summer were similar to those in the bloom. We will address these features in future work related to nutrients and oxygen levels in the water column.
Sediment sources of VOIs in subpolar and polar coastal oceans
We determined the total concentrations (pmol L–1) of VOIs and their percent compositions in the water column and sediment of the study area (Fig. 3). The predominant VOIs in the surface sediments were C2H5I (65–83%) and CH3I (12–29%) at all stations, and the total VOI concentrations (125–252 pmol L–1) in the surface sediments (0–1 cm) were several times higher than those in the bottom water. The total VOI concentrations (37–104 pmol L–1) were lower in the deeper sediment (1–5 cm) than in the surface sediments, and C2H5I still predominated among the four VOIs. As in Funka Bay, we attributed the elevated C2H5I and CH3I in the sediment and bottom water in the Bering and Chukchi Seas to diatom aggregates settled on the sea floor. It has been confirmed that diatoms occupied the great part of phytoplankton communities in the Bering and Chukchi Seas27. At the Chukchi Sea station located at downstream of the Bering Strait, where vertical mixing was well developed throughout the water column, the high ratio (32%) of C2H5I was found in the total water column. We considered that the sediment source VOIs were distributed throughout the water column by mixing and have an impact on VOIs emission to the air at the place.

a Proportional composition (%) of VOIs and their total concentrations (pmol L–1) in the water column and sediment in Chukchi Sea. b Northern Bering Sea. c Bering Sea. d Funka bay. Proportion of CH3I, C2H5I, CH2ClI, and CH2I2 are shown in boxes with gray, white, shade, and black, respectively. The water column was divided into two layers: near-bottom water and the upper water column. The values for the northern Bering Sea and Bering Sea are averages for each area, and the values for Funka Bay are total averages for the time-series observations. Water depths of B (−5 m) and B (−0.3 m) are depths 5 m and 0.3 m above the bottom, respectively.
From microscopic images, several types of centric diatoms and pennate diatoms were found to occupy the phytoplankton community of the plankton net sample collected in the bloom of Funka Bay. We considered that diatom aggregates consisting of various species produce C2H5I and CH3I in darkness. From the underwater video recordings taken at the observation sites (see Supplementary Movie 1–4), we could see many invertebrates covering the sea floor of the Bering Sea at stations 4 and 14, but there were far fewer animals on the sea floor at the other stations. C2H5I predominated in the sediment whether animals were abundant or not on the sea floor. We concluded that surface sediment was an important source of C2H5I and CH3I to the bottom water of global subarctic coastal oceans where diatoms flourish and their impact on VOIs emission to the air was limited in well mixed areas.
VOI production from diatom aggregates in darkness
We confirmed VOI production from diatom aggregates by conducting bottle incubations in darkness using diatom samples collected from Funka Bay during the bloom in 2019. This production is visible in the time-series of the amounts of C2H5I and CH3I collected from the incubation bottles and their total emissions (Fig. 4). The peak amounts of both C2H5I and CH3I were collected on day 3 under oxic conditions and on day 7 under anoxic conditions. The total emissions of C2H5I (anoxic, 27 pmol; oxic, 5.7 pmol) were much greater than those of CH3I (anoxic, 1.6 pmol; oxic, 2.4 pmol). The incubation results are provided in Supplementary Table S1a, b. Our result that C2H5I had the highest production among the VOIs is consistent with a previous incubation study18. The VOI emissions (pmol) per unit chlorophyll-a (mg) were 6.8 pmol (mg chl-a)−1 for C2H5I and 2.8 pmol (mg chl-a)−1 for CH3I under oxic conditions. Iodide addition (+8 μmol L−1) to the incubation bottles resulted in a 7-fold increase in C2H5I production and a 12-fold increase in CH3I production under oxic conditions (Supplementary Table S1c). The iodide addition under anoxic condition resulted in further increases in the productions (Supplementary Table S1d). The temperature (8 °C) of dark incubation was set to be an annual maximum of Funka Bay water at the bottom. Thus, the production rates obtained from the experiments would be a maximum if there is a temperature dependence on the productions associated with biological activity. Note that the dark incubations using diatom aggregates collected in Funka Bay were not sterile, thus the results of VOIs production mean net production including both production and biological loss.

a Collected amount and total emission of C2H5I from the dark incubation bottle. b CH3I. Anoxic condition with N2 supply and oxic condition with air supply are shown in gray square plots and black circle plots, respectively. VOIs were collected from the head-space of diatom incubation bottles in darkness and their total emissions over 42 days. These emissions were collected by flushing with either N2 (anoxic conditions) or air (oxic conditions) as carrier gases. The plots and error bars are averages and ranges of incubation results of the two bottles (n = 2), respectively. Many of error bars are behind the symbols. The incubation results are shown in Supplementary Tables S1–S3.
We also conducted dark incubations using a culture of the diatom Thalassiosira nordenskioeldii, which predominates in the spring bloom in Funka Bay28 (Supplementary Table S2). We started with a sterile culture grown in f/2 medium in the light with added iodide (I−). The diatom cultures with I− produced C2H5I and CH3I when moved to dark conditions. In contrast, iodate (IO3−) addition did not result in remarkable C2H5I or CH3I production. Clearly, I− was necessary for production of the two VOIs. We concluded that both intracellular I− absorbed earlier during their growth and extracellular I− dissolved in seawater could be used for VOI production in darkness. Although bacterial activity would promote VOI production from diatom aggregates in darkness18, our results indicate that bacterial activity is not necessary.
Calculation of VOI production in the sediment of Funka Bay
Primary production in Funka Bay during the spring bloom was 25–73 g-C m–2[23], and the carbon content (μg-C) per unit chlorophyll-a (μg chl-a) of diatoms during the bloom was 11.6 μg-C (μg Chl-a)−1[28]. If all diatoms produced in the bloom had settled to the sea floor, the productions of C2H5I (11–33 nmol m–2) and CH3I (5–14 nmol m–2) on the sea floor could occur applying the emission rates of 6.8 pmol (mg chl-a)–1 for C2H5I and 2.8 pmol (mg chl-a)–1 for CH3I under oxic conditions with no I− addition. Note that the calculated emissions mean net production including production and biological loss. If the emitted C2H5I and CH3I were supplied to the bottom water (80–96 m), the increases of bottom water concentrations were 0.92–2.7 pmol L–1 for C2H5I and 0.38–1.1 pmol L–1 for CH3I. These calculated increases are lower than the actual increases of C2H5I (4.3–4.4 pmol L–1) and CH3I (3.0–5.0 pmol L–1) in bottom water between the middle of March and April of both years. Actually, high levels of C2H5I and CH3I remained in the sediment and bottom water until August. As we have discussed a possibility of biological loss in water column near the bottom, the natural increases involved impacts of production from diatom aggregates and biological loss.
If productions of C2H5I and CH3I from diatom aggregates depend only on an amount of diatom settled on the sea floor, the concentrations of C2H5I and CH3I in the sediment surface in 2019, where the considerably high concentration of chl-a (maximum of 29.7 μg L–1) was found in the bloom 2019 compared with the bloom 2018 (maximum of 12.5 μg L–1) (Fig. 2a), should have become higher than 2018. However, C2H5I and CH3I concentrations in the sediment surface in 2019 were lower than 2018 (Fig. 2b, c). Since I− addition stimulated the productions of C2H5I and CH3I from incubation experiment, we considered that the productions of C2H5I and CH3I in the sediment surface were affected by both the amount of diatom aggregates settled on the sea floor and the supply of iodide (I−) to the aggregates. Moreover, the concentrations of C2H5I and CH3I in the sediment deeper than 1 cm depth was only 20% of the sediment surface (0–1 cm) where the sediment source C2H5I and CH3I were produced; and thus, we considered that the lower concentrations in the deeper sediment were owing to biological (probably bacterial) loss. If the biological losses of C2H5I and CH3I are stimulated by their accumulations in the sediment, the concentrations on the sediment surface should vary dramatically. We suggest that the large variability of C2H5I and CH3I in the sediment surface is caused by their productions and biological losses in the sediment; productions are stimulated by supplies of diatom aggregates settled from the surface and iodide from the deeper sediment as discussed in the next paragraph; biological losses are stimulated by their accumulations in the sediment.
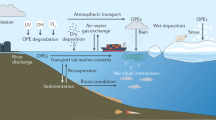
We conclude that the production of VOIs from diatom aggregates in the sediment would have been sustained for several months after the spring bloom by using extracellular I− in the surrounding seawater. Normally, the I−concentration in deep water is much lower than that of IO3−; however, I− could be supplied from deeper sediment in which IO3− is reduced to I− under anoxic conditions29. We have confirmed that I− concentrations in bottom water obviously increased from March to June 2017 in Funka Bay, which were coincident with decrease in IO3− concentrations (Supplementary Fig. S3; unpublished data). The I− and IO3− concentrations were measured according to the stripping voltammetry method30. We considered that continuous supply of I-, which was derived from IO3− reduction in anoxic deeper sediment, to diatom aggregates settled on the sea floor sustained the VOIs productions until August. Because diatoms assimilate iodide faster than iodate during their growth31, we thought that iodine cycle in coastal area is driven by iodide accumulation by phytoplankton. A small fraction of accumulated I− in diatom cell is used for the VOIs productions. We prepared a diagram representing C2H5I emission from diatom aggregates in dark bottom sediment in relation to the iodine cycle between the water column and sediment (Fig. 5).

Iodine cycle between water column and sediment is shown in relation to C2H5I emissions from dark bottom sediment. Iodide in surface seawater is accumulated in phytoplankton cell during their growth (1); the phytoplankton aggregations sink to sea floor (2); IO3− in the deeper sediment is reduced to I− (3); C2H5I and CH3I are produced from the sedimented aggregations using intercellular I− (4) and extracellular I− supplied from deeper sediment (5); large fraction of sediment source I− is supplied to bottom water (6); sediment source C2H5I and CH3I are supplied to the bottom water and lost by microbial activity (7). The pathways of iodine cycle are indicated as arrows.
Our results strongly support the idea that phytodetritus (e.g. diatom aggregates) on the sea floor is a hotspot of VOI production in the ocean18. We concluded that the impact of the sediment source VOIs is limited in the middle layer and bottom layer waters in stratified coastal oceans because of biological losses below the surface layer. However, it is possible that considerable amounts of sediment source VOIs are supplied to the ocean surface and emitted to the air in vertically well-mixed coastal areas, such as more coastal areas and fast-flowing straits. Further research on the sediment source VOIs in subtropical and tropical coastal oceans and more coastal areas of global ocean is needed to evaluate their atmospheric impacts.
Methods
Shipboard observations
Shipboard observations were conducted over the Bering Sea and Chukchi Sea continental shelves from the Training Vessel (T/V) Oshoro-maru in July of 2017 and 2018. Time-series observations in Funka Bay, Hokkaido, Japan, were conducted from the T/V Ushio-maru 17 times from March 2018 to December 2019. Both vessels were operated by the Faculty of Fisheries Sciences, Hokkaido University. Figure 1 shows the locations of the sampling stations. The sea floor at the four stations in the Bering Sea were videotaped during the observations (see Supplementary Movie 1–4). The sea floor at the station 30 of the Funka Bay and observation scenes were recorded in movie files and photos (see Supplementary Movie 5–8 and Supplementary Fig. S5–S16).
Sediment core sampling
Sediment core samples were collected in polycarbonate core tubes (inner diameter 82 mm; length 600 mm) attached to a gravity-type core sampler (Rigo Co. Ltd., Saitama, Japan) for Funka Bay and a multiple core sampler (Rigo Co. Ltd.) for the Bering and Chukchi Seas. The sea-floor depths in the Bering and Chukchi Seas were 40–50 m, and the depth of Funka Bay was 96 m. All sea floors were in darkness below the euphotic zone.
The sediment cores were cut into sections at intervals of 1 cm from the surface (sea floor, 0 cm) to a depth of 5 cm. Each sediment section was collected in a polyethylene bag. The pore water from each sediment section was collected by using a syringe and pulling the water through a porous ceramic filter cup using a soil water sampler (DIK-8393, Daiki Rica Kogyo Co. Ltd., Saitama, Japan) within two hours of the sediment core collection. The pore-water sample in the syringe was filtered into a glass bottle (32 mL) using a syringe filter with a pore size of 0.22 µm. To arrest microbial activity, 50 μL of saturated mercuric chloride (HgCl2) solution was added to the bottle. The bottle was crimp-sealed with 0.5 mL headspace using a butyl rubber septum and an aluminum cap. The sample bottles containing pore water were kept in a refrigerator at 4 °C until pre-treatment for VOI analysis, typically within a month.
Seawater sampling
Seawater samples were collected in Niskin bottles attached to a rosette multi-sampler along with a conductivity-temperature-depth (CTD) probe (SBE 19 plus, Sea-Bird Electronics, Inc., Bellevue, WA, USA). Surface water was collected with a plastic bucket. Bottom waters were collected 1 m above the sea floor using a Van Dorn sampling bottle or a Niskin bottle, and 0.3 m above the sea floor from the sediment-core tubes. The seawater sampling depths were 0, 5, 10, 20, 30, 40, 50, 60, 65, 70, 75, 80, 85, 95 (+1 m above the sea floor), and 95.7 m (+0.3 m above the sea floor) in Funka Bay, and 0, 10, 20, 30, 40, and 50 m depths, and +1 m and +0.3 m above the sea floor, in the Chukchi and Bering Seas. Aliquots of seawater samples (125 mL) were collected into dark glass bottles and approximately 250 mL of seawater was allowed to overflow the bottles. Saturated HgCl2 solution (100 μL) was added to the bottles. The bottles were sealed with a butyl rubber septum and an aluminum cap and kept in a refrigerator until pre-treatment, typically within a month.
Analytical procedures
Dissolved VOIs were collected from the pore water and seawater samples by a purge-and-trap method. The total volume of seawater (125 or 32 mL) in the sample bottle was introduced into a purge vessel by a high-purity nitrogen carrier at 20 mL min−1 for 7 min, and the dissolved gases were consecutively purged by bubbling with nitrogen at 65 mL min−1 for 35 min. The purged gas sample was collected in a cold-trap containing adsorption resin (Tenax TA, 10 mg) cooled to −90 °C. The cold-trap samples were stored in a freezer (−30 °C) until analysis by gas chromatography–mass spectrometry (GC–MS). The purge efficiencies for CH3I, C2H5I, CH2ClI and CH2I2 were 94%, 95%, 83%, and 66%, respectively. Concentrated VOIs in the cold-trap were thermally desorbed and transferred to an automated pre-concentration GC–MS analytical system (capillary column: Porabond Q, 0.32 mm, 50 m; GC–MS: Agilent 5973, 6890). A gravimetrically prepared standard gas (Taiyo Nissan, Inc., Tokyo) in a gas cylinder containing CH3I, C2H5I and other halocarbons at concentrations of 10 ppb (dry air mole fraction) was analyzed with the preconcentration-GC-MS system every measurement date. The concentrations in the cylinder were calibrated by measuring a methanol-based liquid standard containing these compounds every six months. The methanol-based liquid standard was injected into pure water, and then, the water-based standard was measured by purge and trap / GC-MS system. The analytical precisions of CH3I and C2H5I in water sample were ±2%. Details of the GC–MS analysis are described elsewhere20. The amounts of CH3I and C2H5I in the sample bottle (32 mL) reduced by 3−7% a month during the sample storage, thus our results include approximately 3–7% of under estimation.
Chl-a concentrations in discrete seawater samples (100 mL) were measured by a fluorometer model 10-AU-005 (Turner Designs, San Jose, CA, USA). Nutrient concentrations (NO3−, NO2–, NH4+, SiO2, and PO43−) in discrete seawater samples were measured by colorimetric methods using a QuAAtro system (BL-Tec, Osaka, Japan).
VOI production from diatom aggregates in darkness in the laboratory
Diatom aggregates were collected from the same location in Funka Bay (St. 30) on 15 March 2019 and 18 March 2021 during massive spring diatom blooms (>15 μg L–1 of chl-a). A plankton net (0.3 m diameter, 100 μm mesh) was towed vertically from 85 m depth to the surface. The diatom aggregates collected in 2019 were suspended in 900 mL of seawater and frozen (−25 °C) in a polyethylene bag until preparation for the incubation experiment. Supplementary Fig. S4 is a schematic showing the preparation protocol for the incubation experiment. An aliquot (9 mL) of defrosted diatom aggregate sample was put in a 200 mL glass bottle. Fluorometric analysis showed that 9 mL of the diatom aggregate contained 0.84 mg chlorophyll-a. Filtered seawater (100 mL) was added to the glass bottle. The diatom aggregates collected by plankton net in 2021 were transferred to the incubation bottle within 2 h after sampling without freezing. A spot sensor for an optical O2 meter (FireSting, PyroScience GmbH, Hubertusstr, Germany) was attached to the inner wall of the incubation bottles to measure the oxygen level in the bottle. The bottle was sealed with a butyl rubber septum and an aluminum cap.
We set up two bottles each for anoxic conditions and oxic conditions. The 12 incubation bottles (A-1—D-3; see Supplementary Table S1a–d) were placed in a dark incubator at 8 °C. Iodide solution was added (+8 μmol L–1) to the bottles C and D. Head-space gas of each bottle was sampled on days 0, 3, 7, 14, 21, 28, 35, and 42 after the start of incubations. Carrier gas (N2 or pure air) was introduced to the bottle through a needle at a flow rate of 20 mL min–1 for 30 min at each sampling time. The head-space gas was collected through another needle. The water in the bottle was kept at 8 °C and magnetically stirred during the gas collection. The water vapor in the gas sample was removed by a glass-tube water trap at −25 °C. The VOIs in the gas sample were collected in a cold trap with adsorption resin (Tenax TA) at −90 °C. The oxygen level in the bottles was measured before and after the head-space gas collection. Analysis for VOIs was the same as for the seawater samples. The collection efficiency of VOIs from the bottles, based on one head-space gas collection, was 28%. The calculation of the total amount of VOIs emission was described in explanatory notes of Supplementary Table S1a–d.
We conducted another dark incubation experiment to examine effect of iodine addition. A sterile culture of diatom, Thalassiosira nordenskioeldii, was grown in Si-rich f/2 medium (NO3−, 700 μmol L−1; PO43−, 26 μmol L–1; Si(OH)4, 75 μmol L–1) with addition of iodide (I−, 8 μmol L–1) or iodate (IO3−, 8 μmol L–1). Then, the cultures were used for the bottle incubation in darkness under oxic condition. We set up two bottles for each no iodine addition, I− addition, and IO3− addition. VOIs were collected from the incubation bottles on days 0 and 11 (Supplementary Table S2).
We also conducted a dark incubation experiment to test if diatom aggregate without prior freezing produces VOIs. We collected diatom aggregates in the Funka-bay by plankton net towing from the depth of 85 m to the surface in spring bloom 2021. The diatom sample was not frozen before the incubation. An aliquot (9 mL) of diatom aggregate sample was put in a 200 mL glass bottle. We prepared 10 incubation bottles. The first VOIs collection from the bottle was conducted after 6 days of the net sampling. The head space of the bottle was collected on days 6, 20, and 26 (Supplementary Table S3). Since we did not collect VOIs on day 0, the initial concentration was not obtained. We confirmed that the diatom aggregates without prior freezing produced CH3I (0.7–2.2 pmol) and C2H5I (11.5–16.5 pmol) for 20 days or 26 days of incubation time.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The datasets generated during the current study are available at https://doi.org/10.5281/zenodo.6905361.
References
Miyake, Y. & Tsunogai, S. Evaporation of iodine from the ocean. J. Geophys. Res. 68, 3989–3993 (1963).
Saiz-Lopez, A. et al. Atmospheric Chemistry of Iodine. Chemical Rev. 112, 1773–1804 (2012).
Koenig, T. K. et al. Quantitative detection of iodine in the stratosphere. PNAS 117, 1860–1866 (2020).
Yokouchi, Y. et al. Distribution of methyl iodide, ethyl iodide, bromoform and dibromomethane over the ocean (east and southeast Asian seas and the western Pacific). J. Geophys. Res. 102, 8805–8809 (1997).
Yokouchi, Y. et al. Diurnal and seasonal variations of iodocarbons (CH2ClI, CH2I2, CH3I, and C2H5I) in the marine atmosphere. J. Geophys. Res. 116, D06301 (2011).
Ooki, A. et al. A global-scale map of isoprene and volatile organic iodine in surface seawater of the Arctic, Northwest Pacific, Indian, and Southern Oceans. J. Geophys. Res. 120, 4108–4128 (2015).
Archer, S. D. et al. Marked seasonality in the concentrations and sea-to-air flux of volatile iodocarbon compounds in the western English Channel. J. Geophys. Res. 112, C08009 (2007).
Jones, C. E. et al. Quantifying the contribution of marine organic gases to atmospheric iodine. Geophys. Res. Lett. 37, L18804 (2010).
Schall, C. et al. Biogenic volatile organoiodine and organobromine compounds released from polar macroalgae. Chemosphere 28, 1315–1324 (1994).
Giese, B. F. et al. Release of volatile iodinated C1–C4 hydrocarbons by marine macroalgae from various climate zones. Environ. Sci. Technol. 33, 2432–2439 (1999).
Manley, S. L. & de la Cuesta, J. L. Methyl iodide production from marine phytoplankton cultures. Limnol. Oceanogr. 42, 142–147 (1997).
Tokarczyk, R. & Moore, R. M. Production of volatile organohalogens by phytoplankton cultures. Geophys. Res. Lett. 21, 285–288 (1994).
Moore, R. M. et al. Bromoperoxidase and iodoperoxidase enzymes and production of halogenated methanes in marine diatom cultures. J. Geophys. Res. 101, 20899–20908 (1996).
Smythe-Wright, D. et al. Methyl iodide production in the ocean: Implications for climate change. Global Biogeochem. Cycles. 20, GB3003 (2006).
Hughes, C. D. et al. Iodomethane production by two important marine cyanobacteria: Prochlorococcus marinus (CCMP 2389) and Synechococcus sp (CCMP 2370). Mar. Chem. 125, 19–25 (2011).
Fuse, H. et al. Production of free and organic iodine by Roseovarius spp. FEMS Microbiol. Lett. 229, 189–194 (2003).
Amachi, S. et al. Isolation of iodide-oxidizing bacteria from iodide-rich natural gas brines and seawaters. Microb. Ecol. 49, 547–557 (2005).
Hughes, C. et al. The production of volatile iodocarbons by biogenic marine aggregates. Limnol. Oceanogr. 53, 867–872 (2008).
Moore, R. M. & Zafiriou, O. C. PHOTOCHEMICAL PRODUCTION OF METHYL-IODIDE IN SEAWATER. J. Geophysical Res. Atmos. 99, 16415–16420 (1994).
Ooki, A. & Yokouchi, Y. Determination of Henry’s law constant of halocarbons in seawater and analysis of sea-to-air flux of iodoethane (C2H5I) in the Indian and Southern oceans based on partial pressure measurements. Geochem. J. 45, e1–e7 (2011).
Ooki, A. et al. Concentration maxima of volatile organic iodine compounds in the bottom layer water and the cold, dense water over the Chukchi Sea in the western Arctic Ocean: possibility of production related to the degradation of organic matter. Biogeosciences 13, 133–145 (2016).
Shimizu, Y. et al. Seasonal variation of volatile organic iodine compounds in the water column of Funka Bay, Hokkaido, Japan. J. Atmos. Chem. 74, 205–225 (2017).
Kudo, I. & Matsunaga, K. Environmental factors affecting the occurrence and production of the spring phytoplankton bloom in Funka bay, Japan. J. Oceanogr. 55, 505–513 (1999).
Isada, T. et al. Influence of hydrography on the spatiotemporal variability of phytoplankton assemblages and primary productivity in Funka Bay and the Tsugaru Strait. Estuarine Coastal Shelf Sci. 188, 199–211 (2017).
Ooki, A. et al. Isoprene production in seawater of Funka Bay, Hokkaido, Japan. J Oceanogr. 75, 485–501 (2019).
Moore, R. M. Methyl halide production and loss rates in sea water from field incubation experiments. Marine Chem. 101, 213–219 (2006).
Giesbrecht, K. E. et al. A decade of summertime measurements of phytoplankton biomass, productivity and assemblage composition in the Pacific Arctic Region from 2006 to 2016. Deep-Sea Res. Part II-Topical Studies Oceanogr. 162, 93–113 (2019).
Ban, S. et al. In situ egg production and hatching success of the marine copepod Pseudocalanus newmani in Funka Bay and adjacent waters off southwestern Hokkaido, Japan: associated to diatom bloom. J. Plankton Res. 22, 907–922 (2000).
Anschutz, P. et al. Interactions between metal oxides and species of nitrogen and iodine in bioturbated marine sediments. Geochimica et Cosmochimica Acta. 64, 2751–2763 (2000).
Campos, M. New approach to evaluating dissolved iodine speciation in natural waters using cathodic stripping voltammetry and a storage study for preserving iodine species. Marine Chem. 57, 107–117 (1997).
de la Cuesta, J. L. & Manley, S. L. Iodine assimilation by marine diatoms and other phytoplankton in nitrate-replete conditions. Limnology Oceanogr. 54, 1653–1664 (2009).
Acknowledgements
We thank the captains and crews of T/V Ushio-maru and T/V Oshoro-maru (Hokkaido University). This research was supported financially by FYs 2018–2019 Research Projects from the Hokusui Society Foundation, Sapporo, Japan, the Japan Society for the Promotion of Science (JSPS) KAKENHI grant numbers 16H02929 and 16H01586, and Arctic Challenge for Sustainability (ArCS) (JPMXD1300000000) / ArCS II projects funded by the Ministry of Education, Culture, Sports, Science, and Technology of Japan (MEXT). Videography in the Bering and Chukchi Seas was supported by KANSO TECHNOS CO., LTD., Osaka, Japan.
Author information
Authors and Affiliations
Contributions
A.O. designed the research, conducted observations and incubation experiment using sterile diatom and diatom aggregation collected in 2021 and wrote the manuscript with contributions from all co-authors. K.M. conducted observations in Funka Bay and Bering/Chukchi Seas and VOIs analysis and supported the data analysis and discussion about VOIs in Funka Bay 2018-2019. F.M. conducted the observations in Funka Bay and incubation experiments using diatom aggregation collected in 2019 and supported the data analysis and discussion about incubation experiment. N.M. conducted the observations in Funka Bay and VOIs analysis and supported the data analysis and discussion about VOIs in Funka Bay 2019. T.H. designed the Oshoro-maru cruise and supported the data analysis about basic parameter in Bering and Chukchi Seas. H.U. designed the Oshoro-maru cruise and supported the data analysis about basic parameter in Bering and Chukchi Seas. Y.N. supported the sterile diatom incubations and the data analysis about the experiment. T.T. designed the Ushio-maru cruise and supported the data analysis about the basic parameter in Funka Bay.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Earth & Environment thanks Stephen Archer and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Clare Davis. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ooki, A., Minamikawa, K., Meng, F. et al. Marine sediment as a likely source of methyl and ethyl iodides in subpolar and polar seas. Commun Earth Environ 3, 180 (2022). https://doi.org/10.1038/s43247-022-00513-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43247-022-00513-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
